2 分子结构
MView可以通过SMILES创建分子结构,也可以通过导入含分子结构的文件导入分子结构。
2.1 创建分子
点击生成结构按钮,在对应文本框中输入分子的SMILES,按回车或单击生成结构,即可生成对应的分子结构。
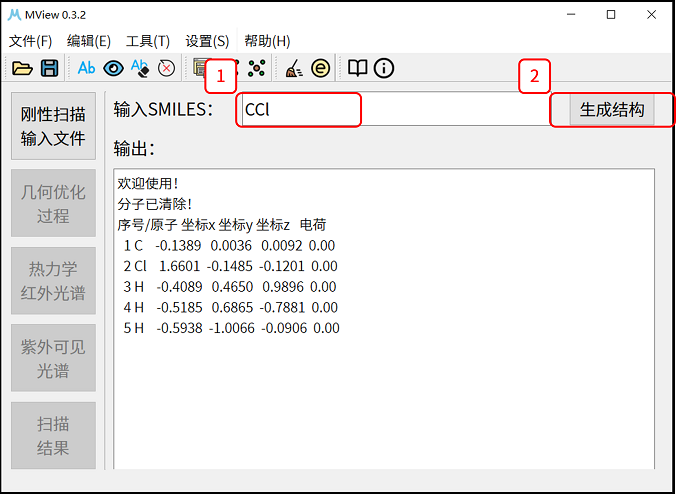
2.2 导入分子
通过将文件拖入MView主窗口,或者点击【打开】按钮、点击菜单栏【文件】-【打开】、键盘输入O+O快捷键并进行选择,均可打开对应的分子结构文件。
支持的输入格式有:
CP2K结构文件(.coord)
CP2K输入文件(.inp)
CP2K振动文件(VIBRATIONS.mol)
Dmol3输出文件(.outmol)
Gaussian检查点文件(.fch或.fchk)
Gaussian输入文件(.gjf)
Gaussian输出文件(.out或.log)
mol文件(.mol或.sdf)
mol2文件(.mol2)
ORCA输入文件(.inp)
ORCA输出文件(.out)
pdb文件(.pdb)
Psi4输入文件
Psi4输出文件
PubChem的JSON文件(.json)
PySCF输出文件(.txt)
xsd文件(.xsd)
xTB输出文件(.log)
xTB输出文件(g98.out)
xyz文件(.xyz)
如果是包含多个结构的文件,将默认读取最后一个结构到MView中。
2.3 查看分子
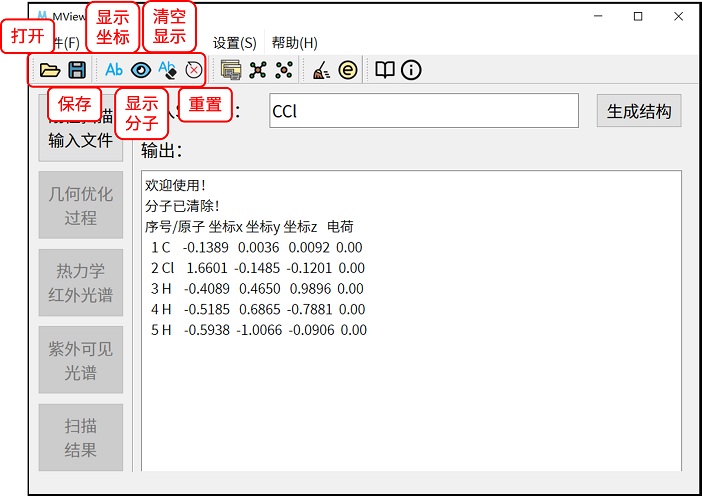
2.3.1 显示分子坐标
在生成或导入分子结构后,输出界面将自动显示分子中各原子的坐标数值,数据最后一列将显示原子电荷值,如导入文件中不存在电荷信息,则最后一列显示0.00。
单击工具栏中【清空显示】按钮能够清除输出窗口显示的文本信息,MView中的分子结构不会被清除。
单击工具栏中【显示坐标】按钮能够重新输出分子的坐标数值。
2.3.2 可视化
MView目前不自带可视化功能,可以借助外接可视化程序的方式实现可视化。
外接好可视化工具后,单击工具栏中的【显示分子】按钮即可调用对应工具显示分子结构。
2.4 删除分子
单击【编辑】-【清除分子】,可以删除MView中的分子,同时出界面不会清空。
单击工具栏中【重置】按钮,可以重置整个MView程序,此时MView中的分子将被删除,且输出界面将会清空。
注意:删除分子是不可逆操作,将会完全丢失程序中的分子信息!
2.5 保存分子
在MView中存在分子结构的情形下,单击工具栏中【保存】按钮,可以将分子数据保存为文件。
支持的输出格式有:
Gaussian直角坐标输入文件(.gjf)
Gaussian内坐标输入文件(.gjf)
Orca输入文件(.inp)
其它常见分子结构文件(.mol,.mol2,.pdb,.xyz)