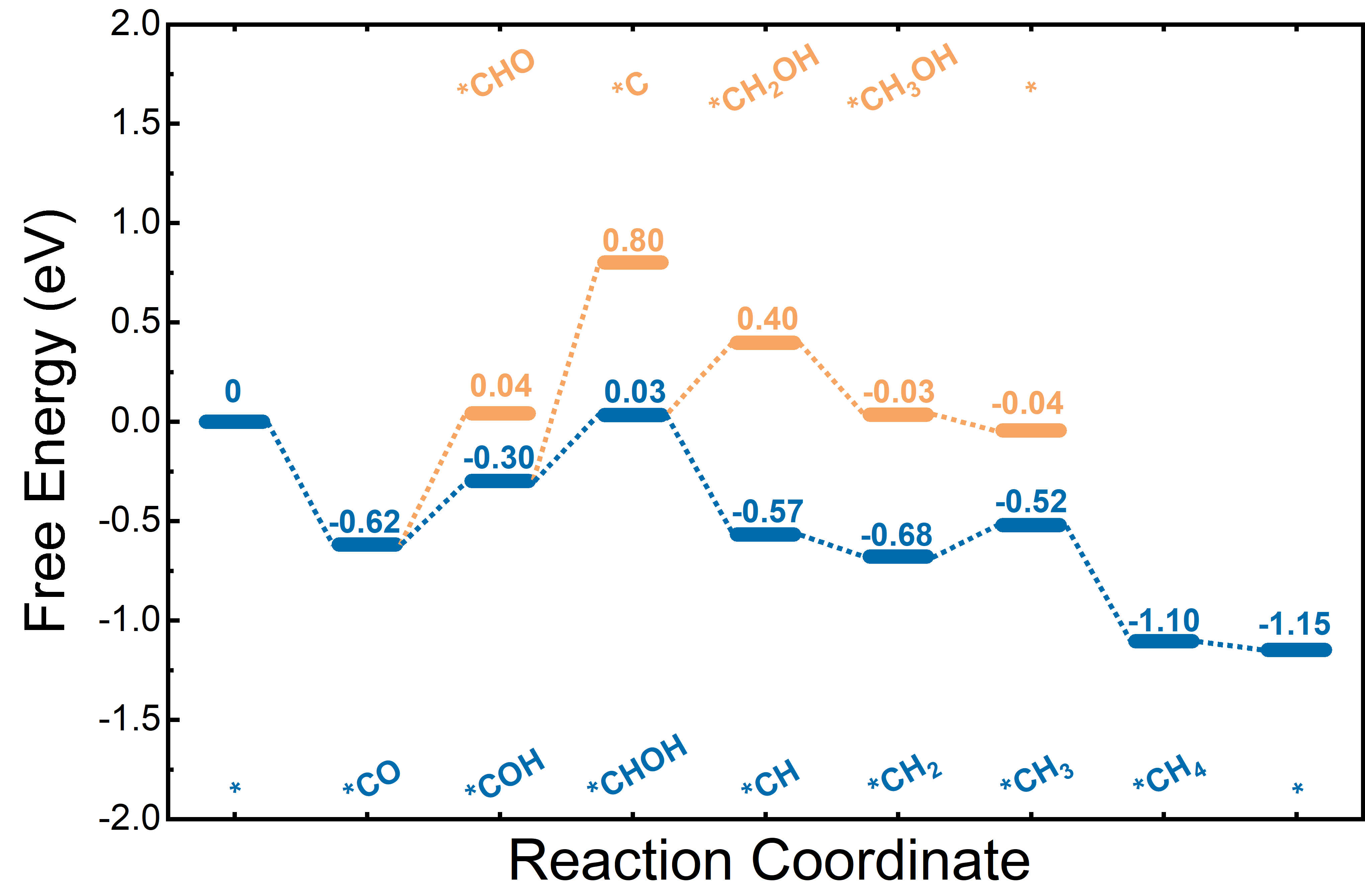
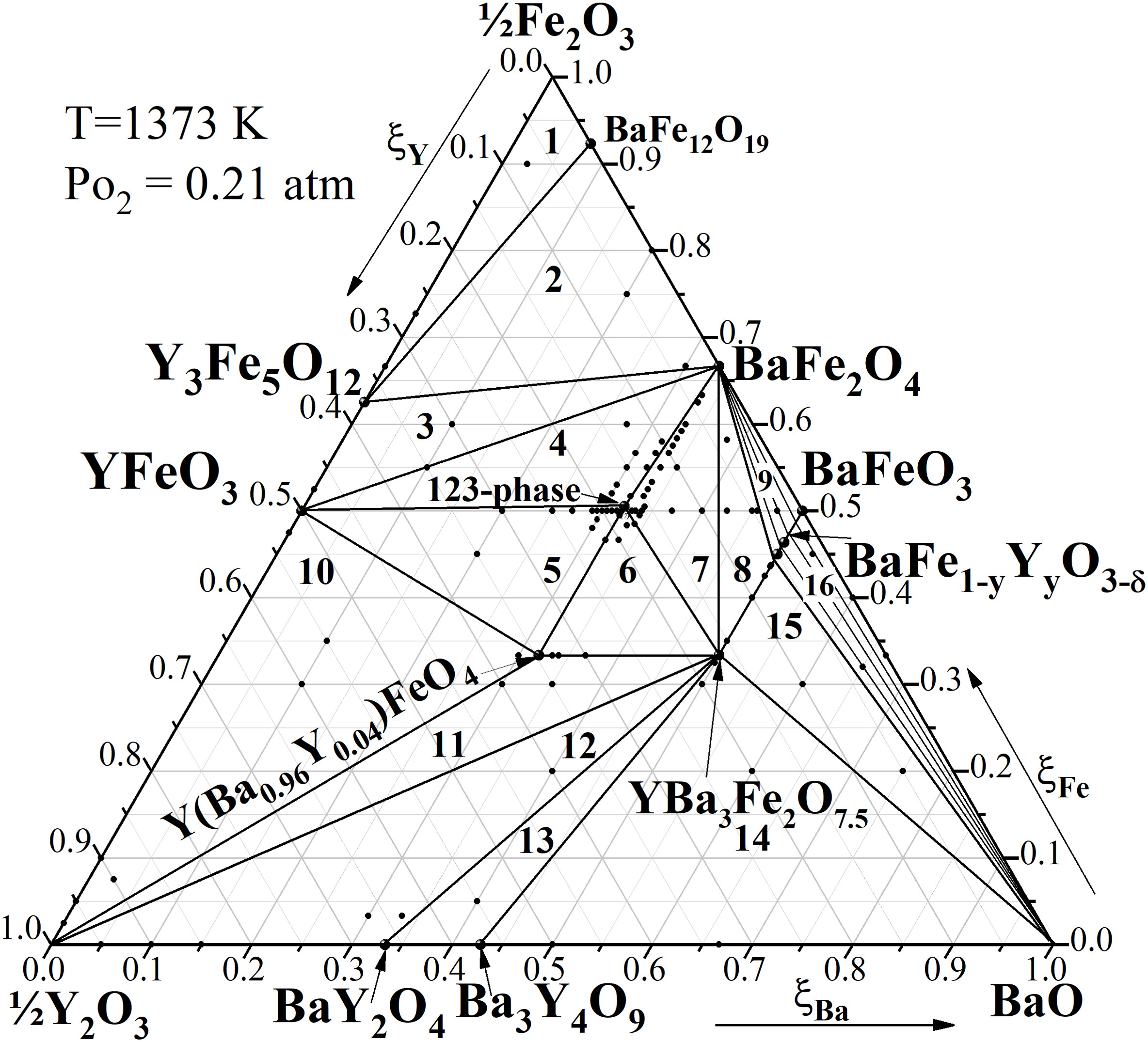
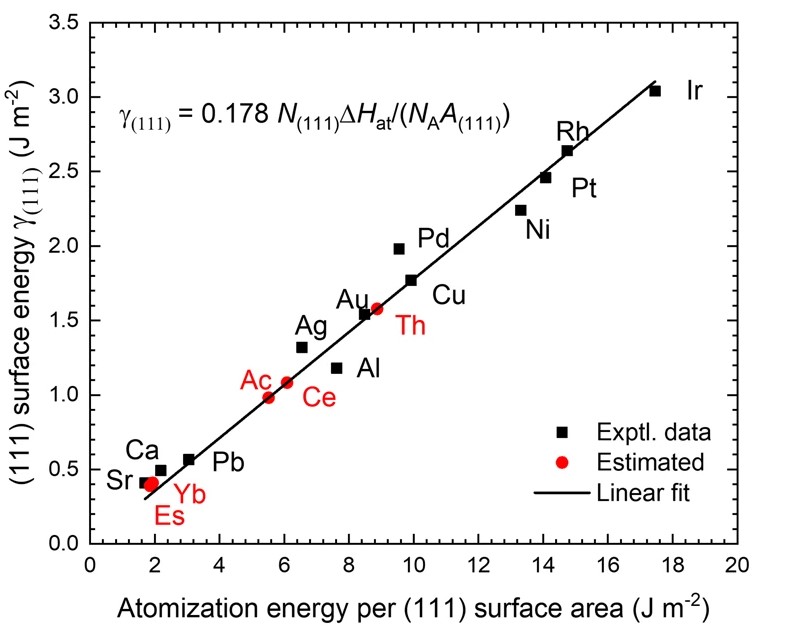
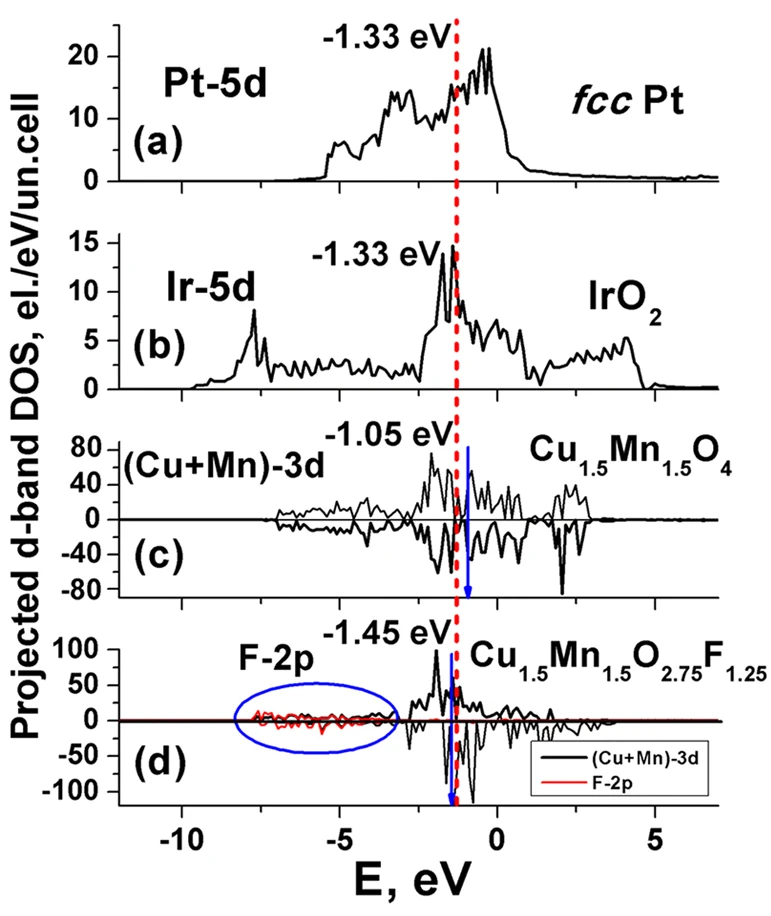
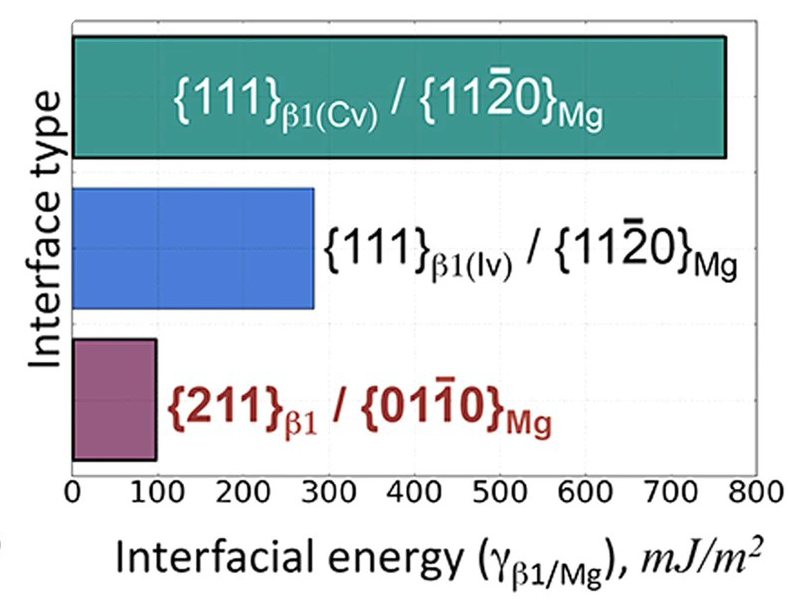
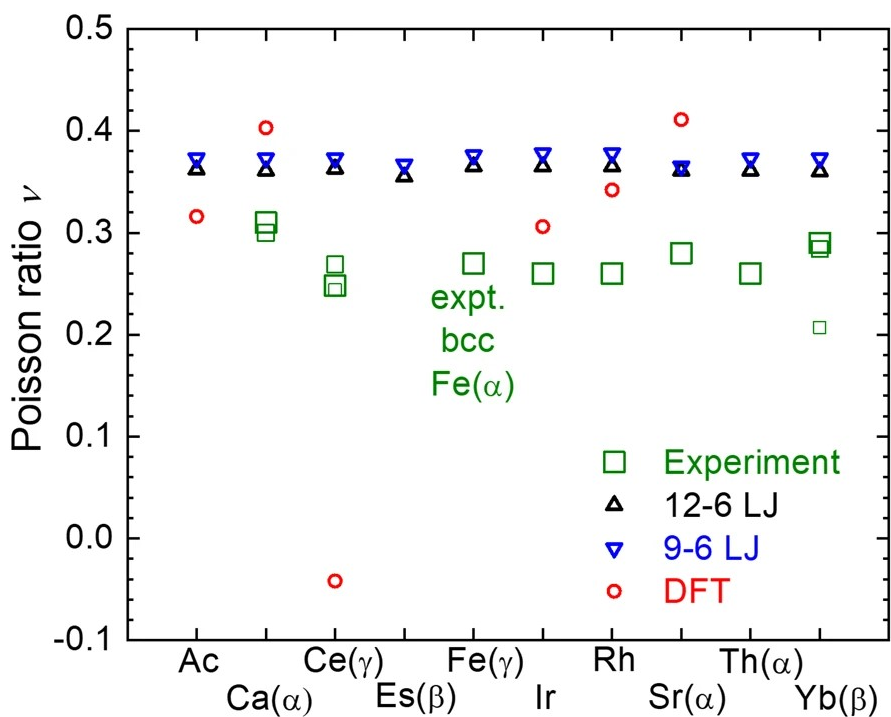
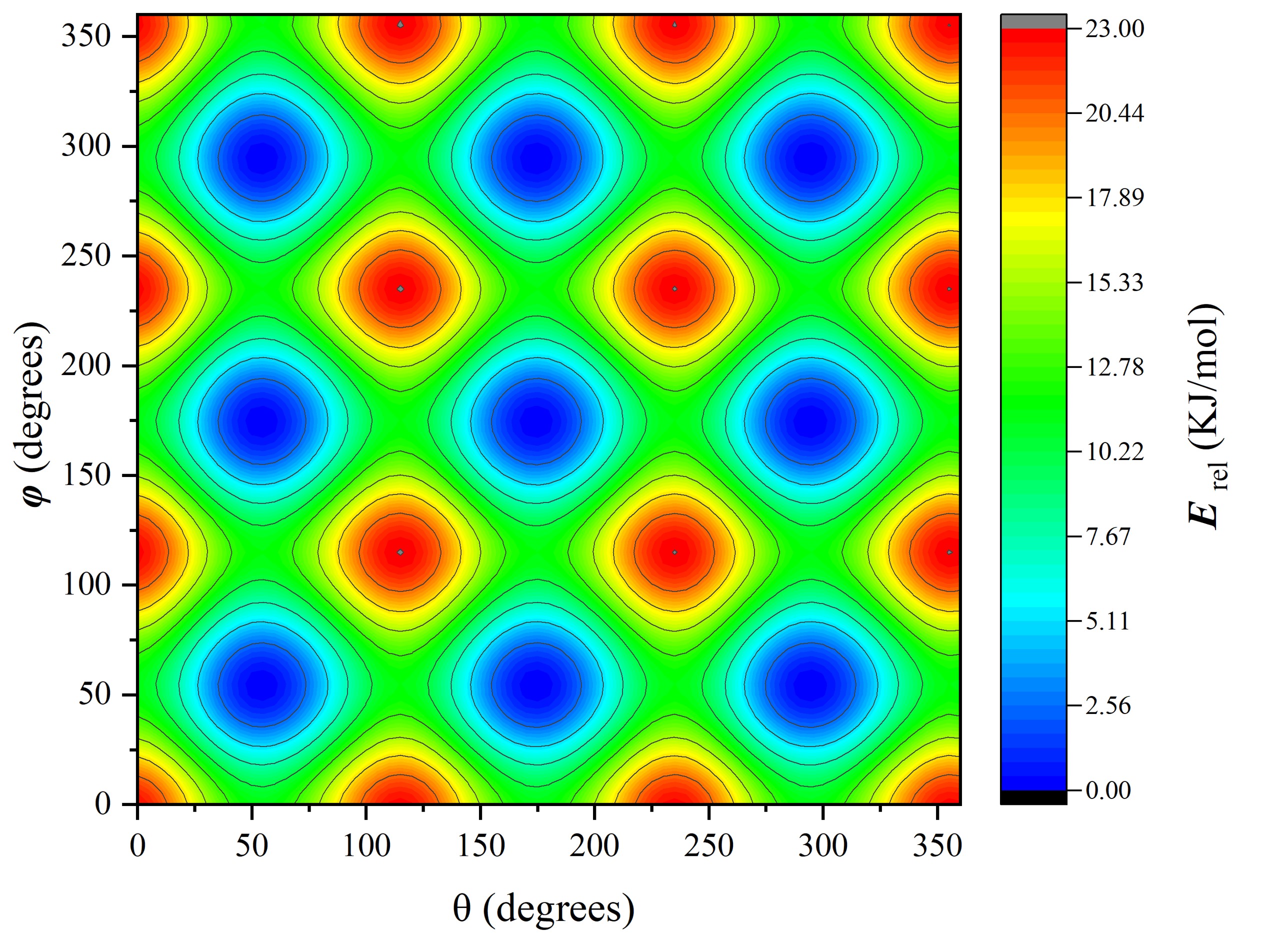
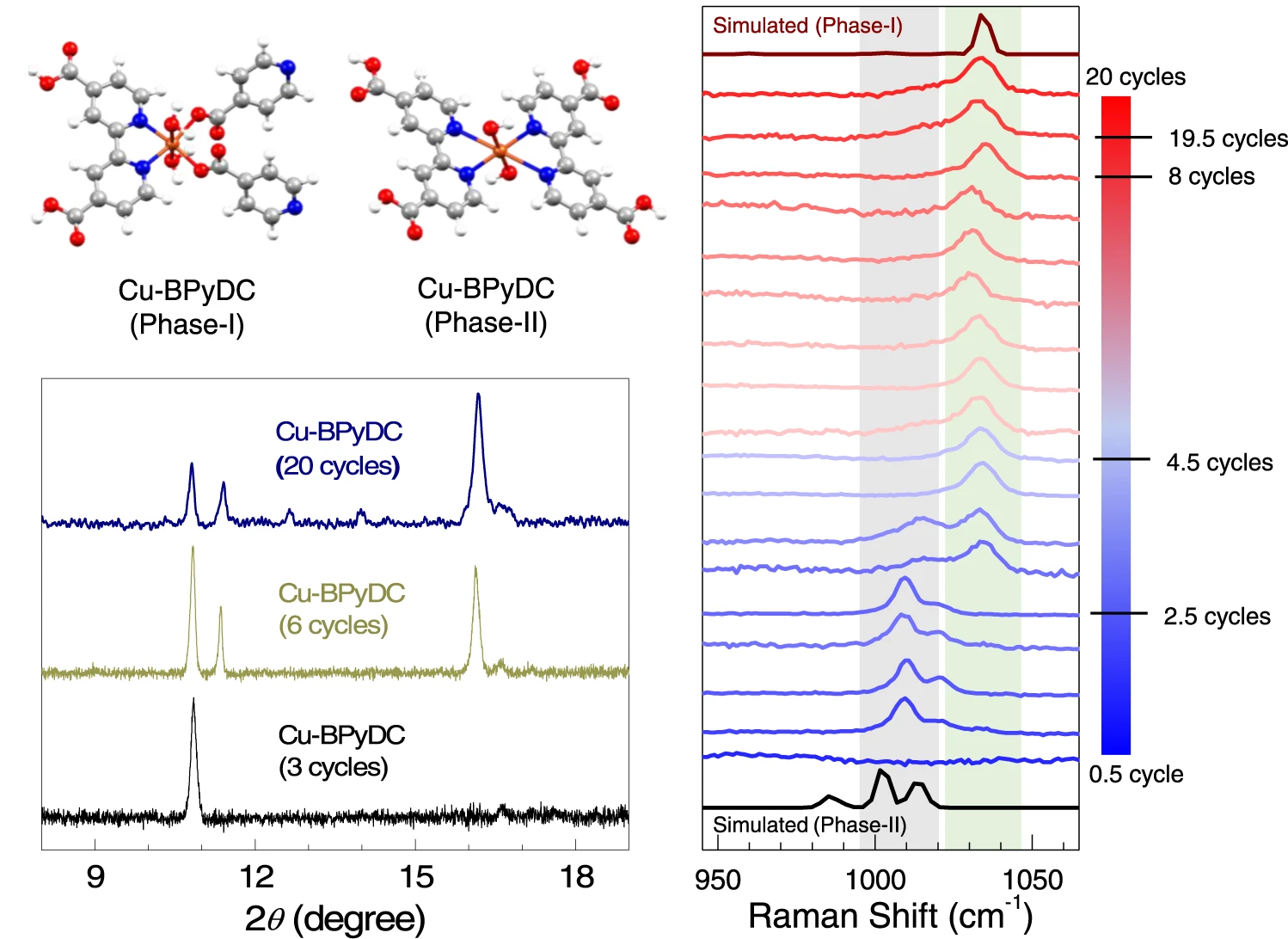
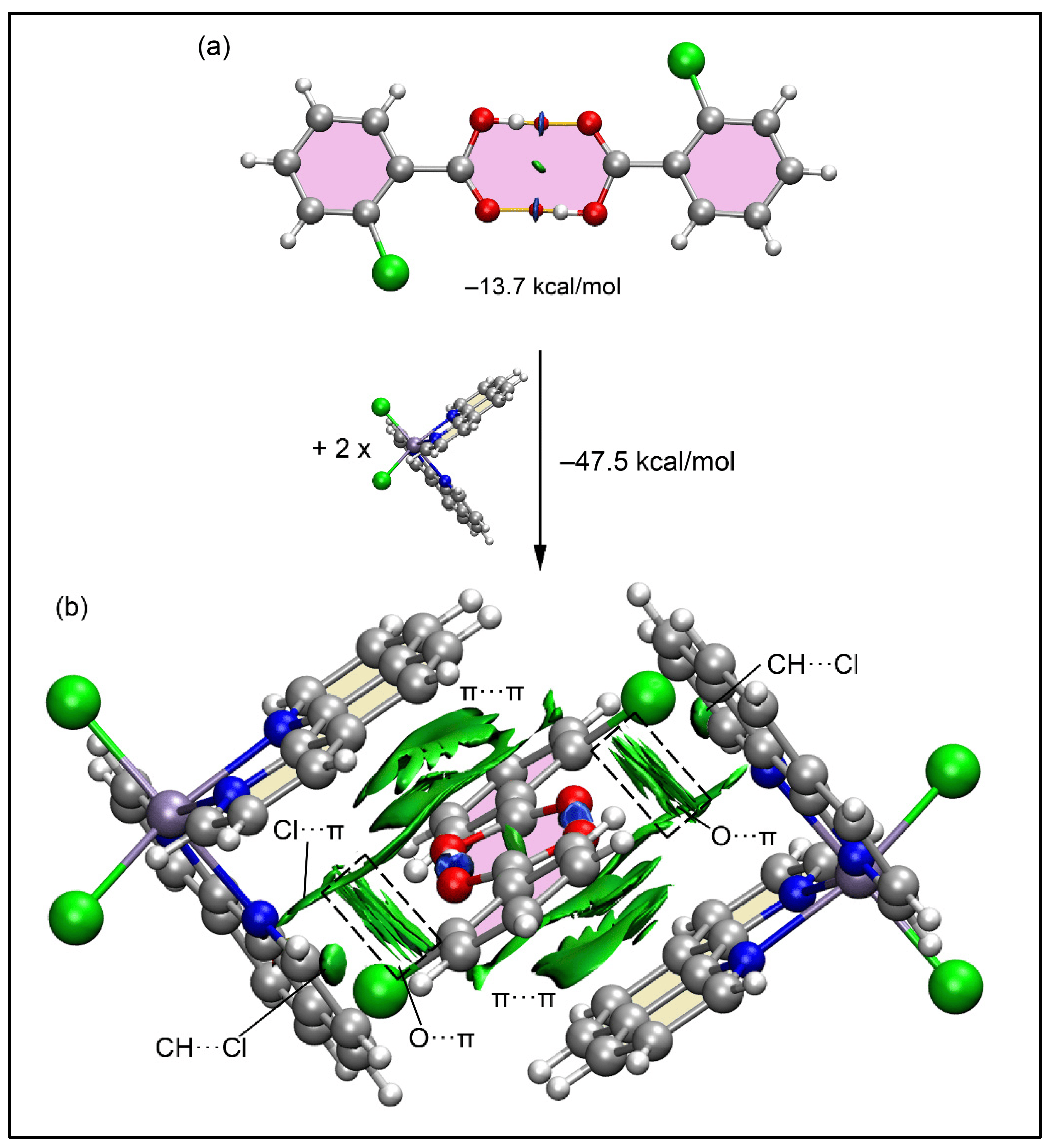
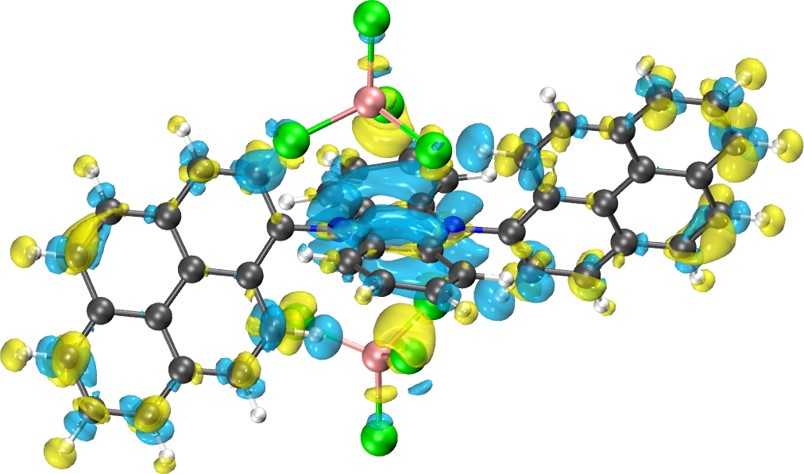
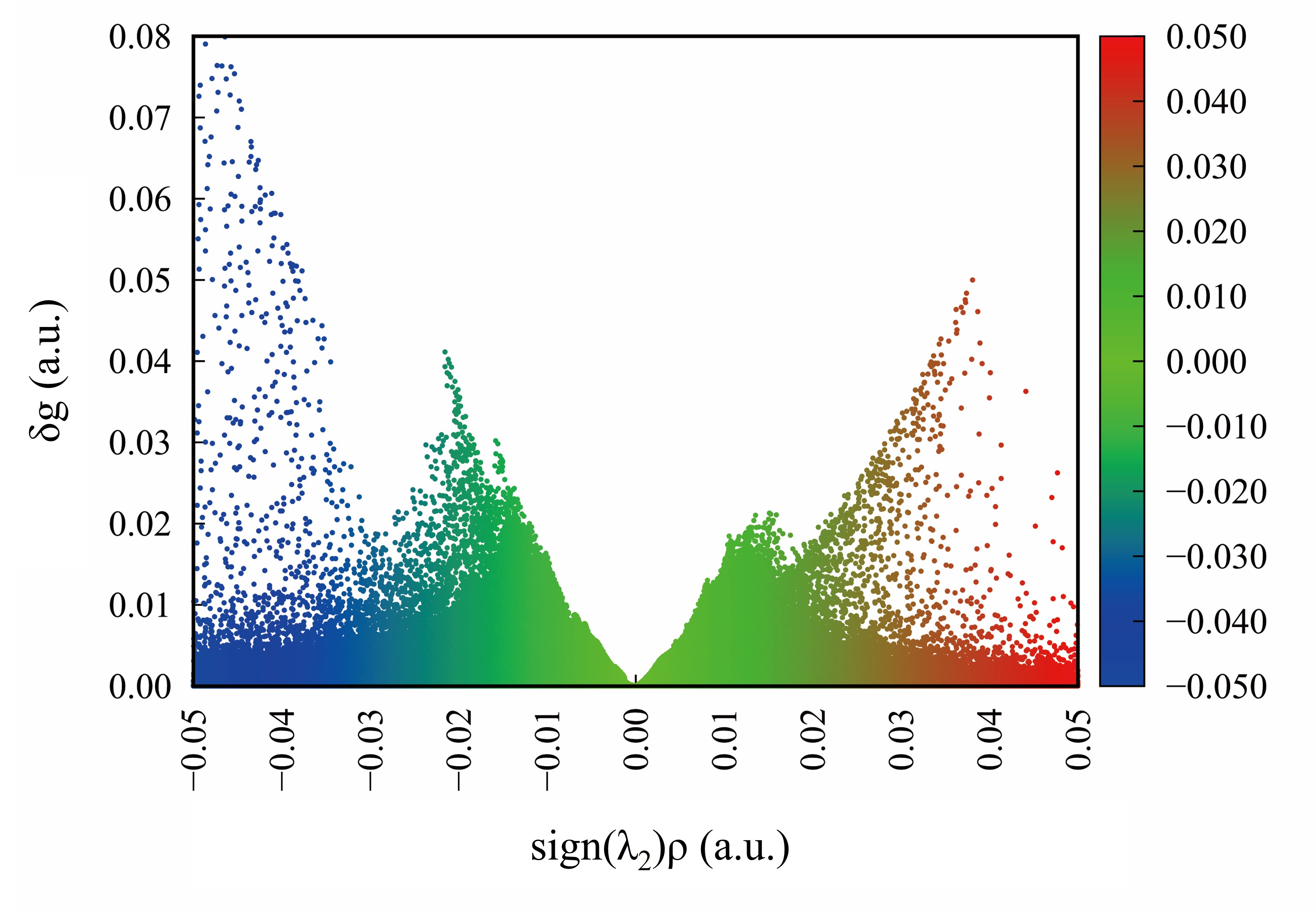
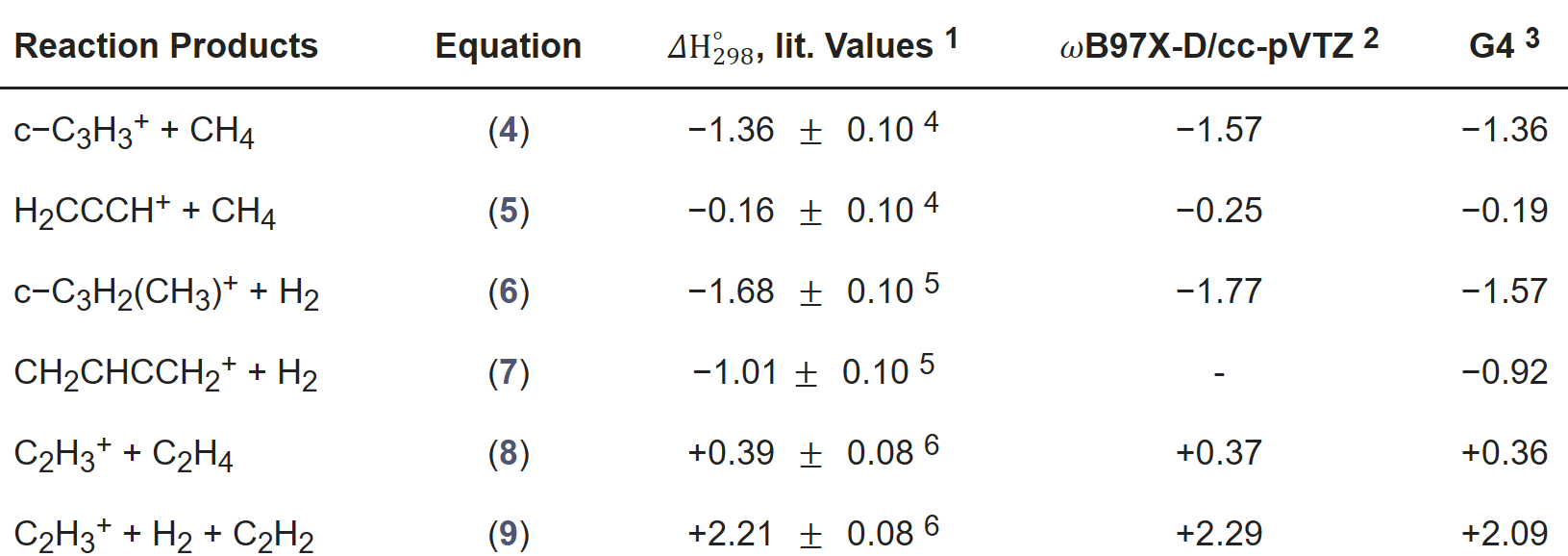
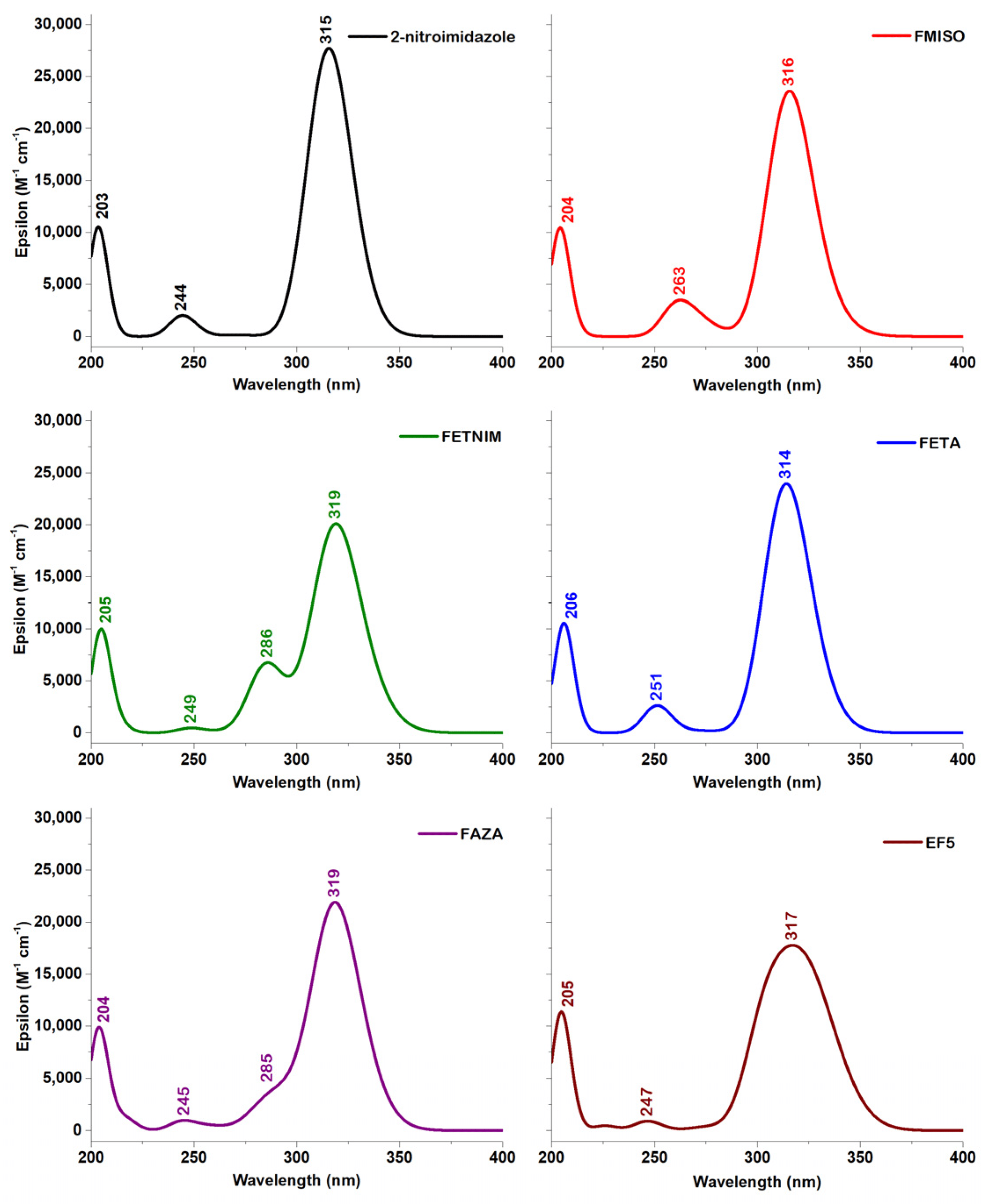
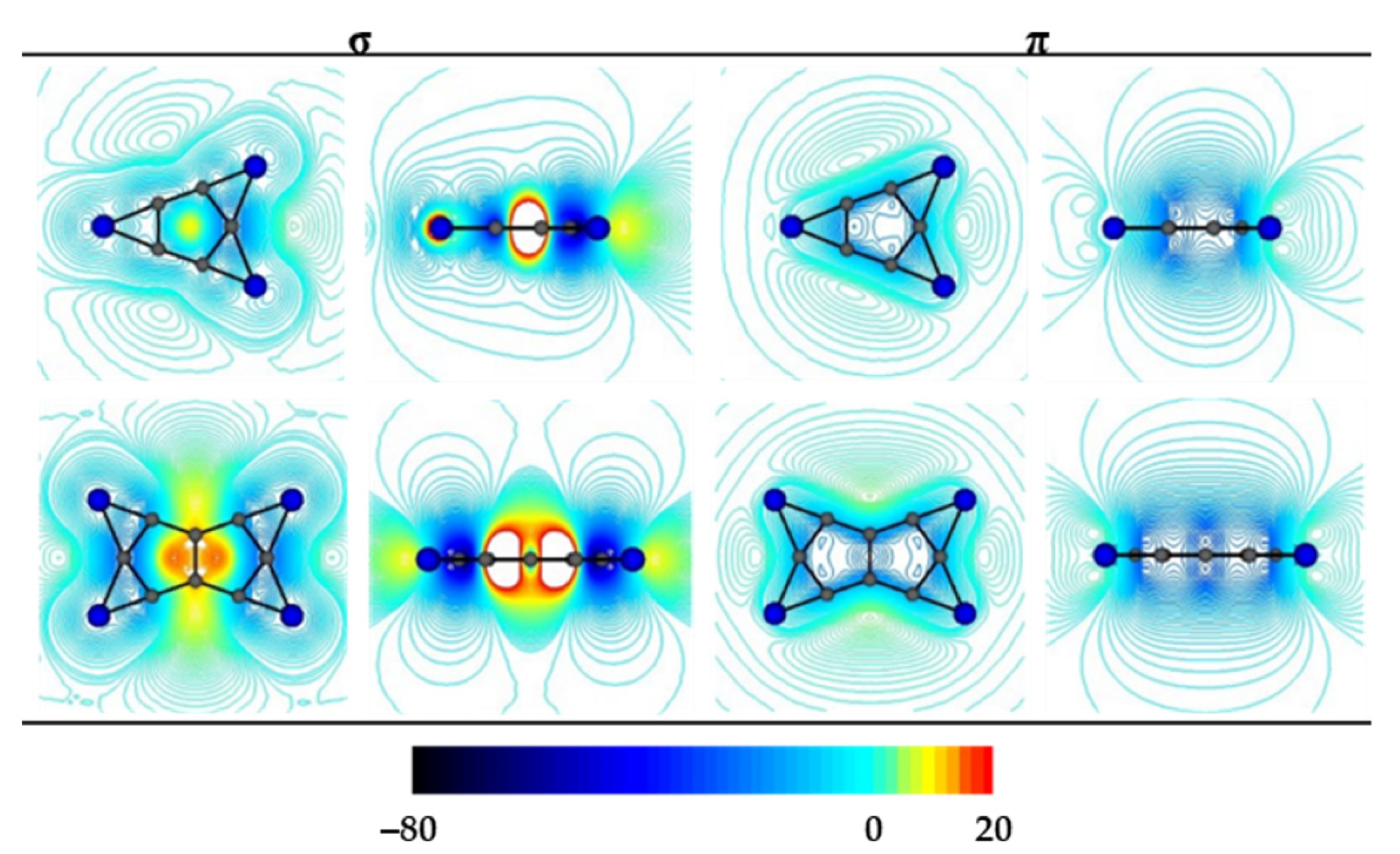
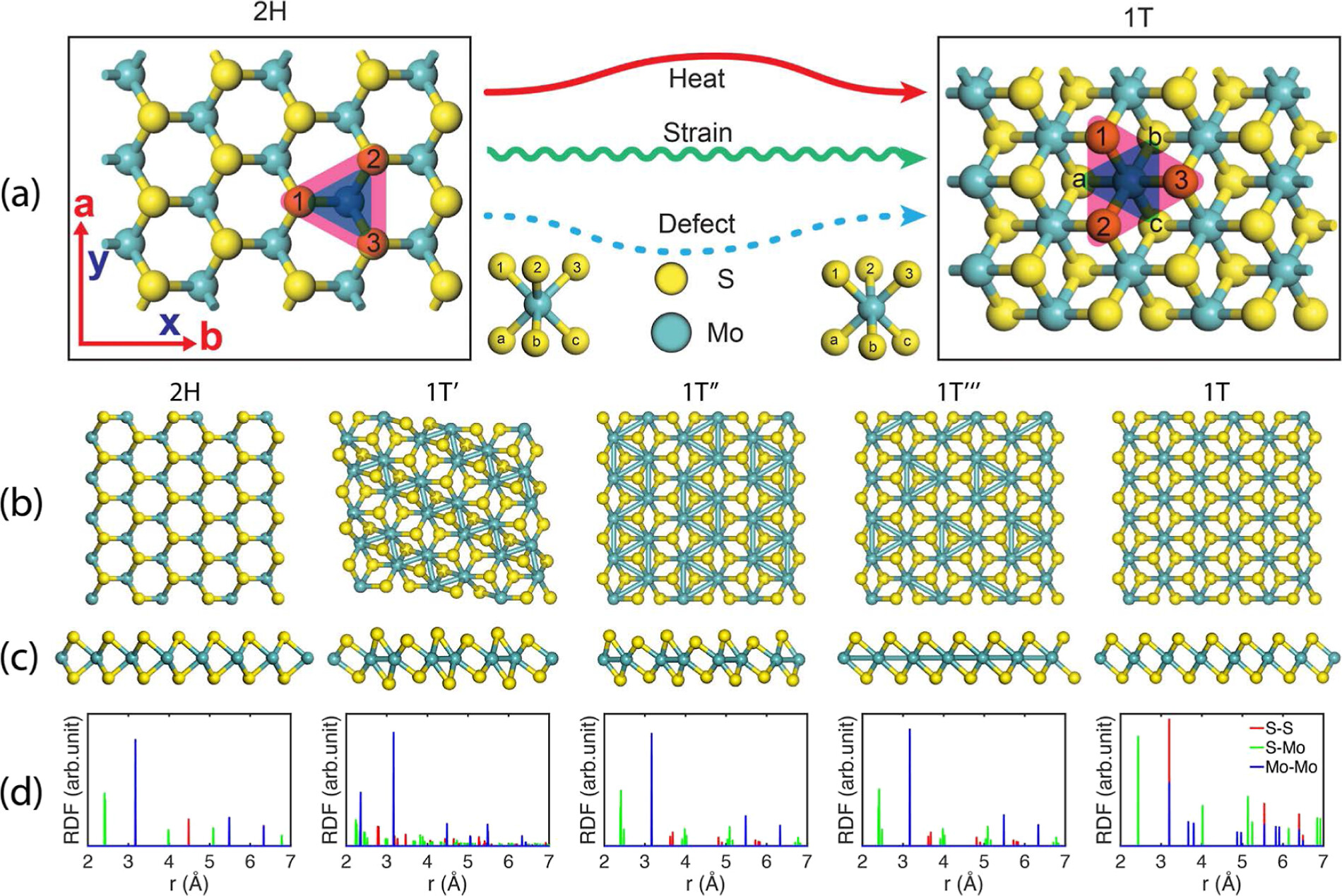
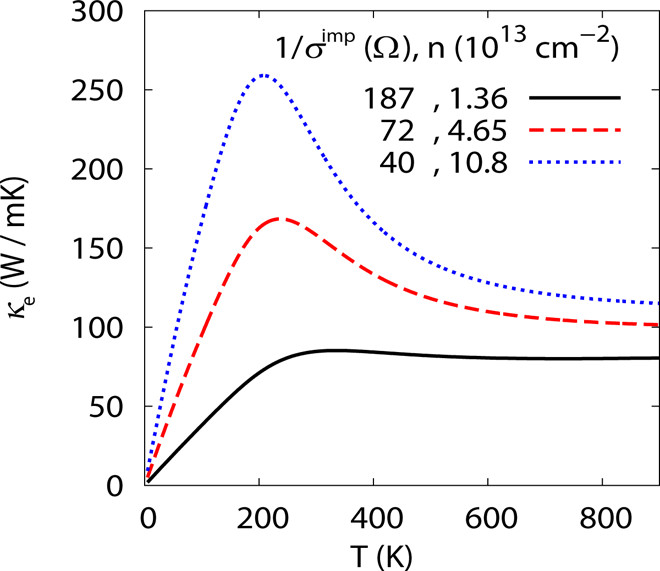
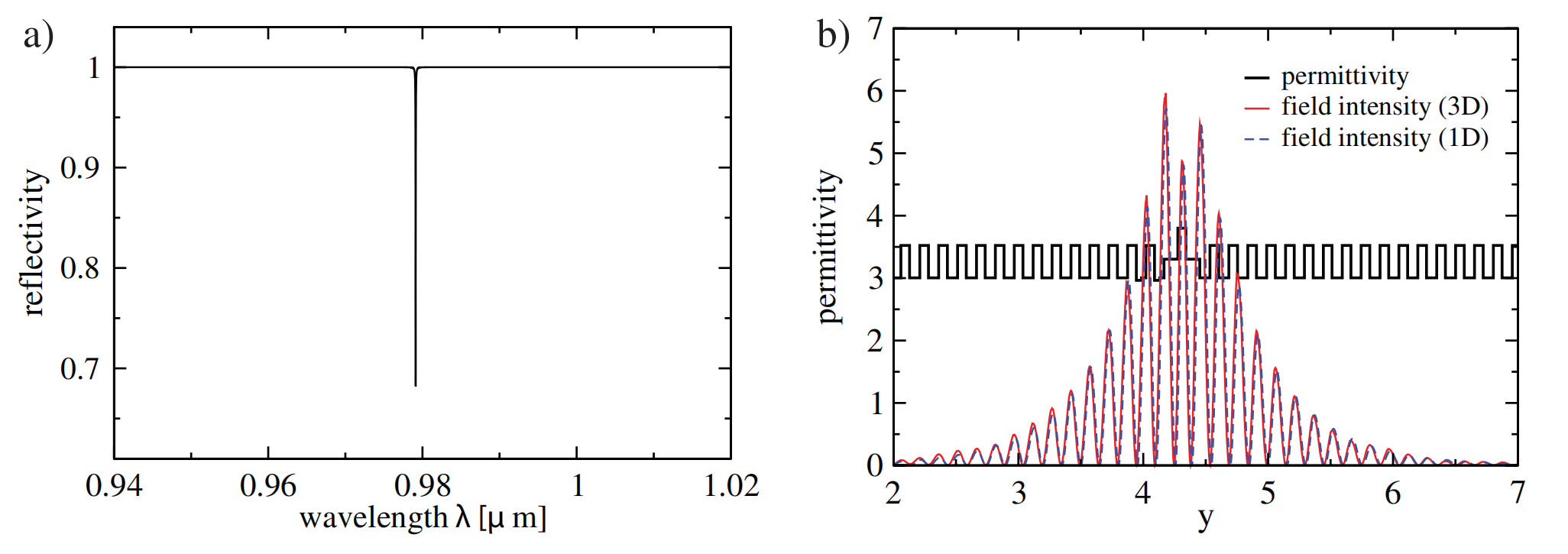
晶格常数、原子坐标、键长、键角、XRD、 电荷密度、态密度、能带,投影态密度、投影能带、COHP、静电势、ELF、功函数、吸附能、结合能、偏析能、界面能、表面能、形成能、差分电荷密度、bader电荷、mulliken电荷、过渡态搜索、OER、ORR、HER、NRR、CO2RR、NO3RR、NORR、SRR、迁移能垒、带边电位匹配、内建电场、 介电函数、折射率、吸收光谱、反射光谱、能量损失函数、声子谱、声子态密度、磁矩、磁化率、自旋密度、弹性常数、弹性模量、体积模量、德拜温度、原子振动频率、压电张量、隐式溶剂模型


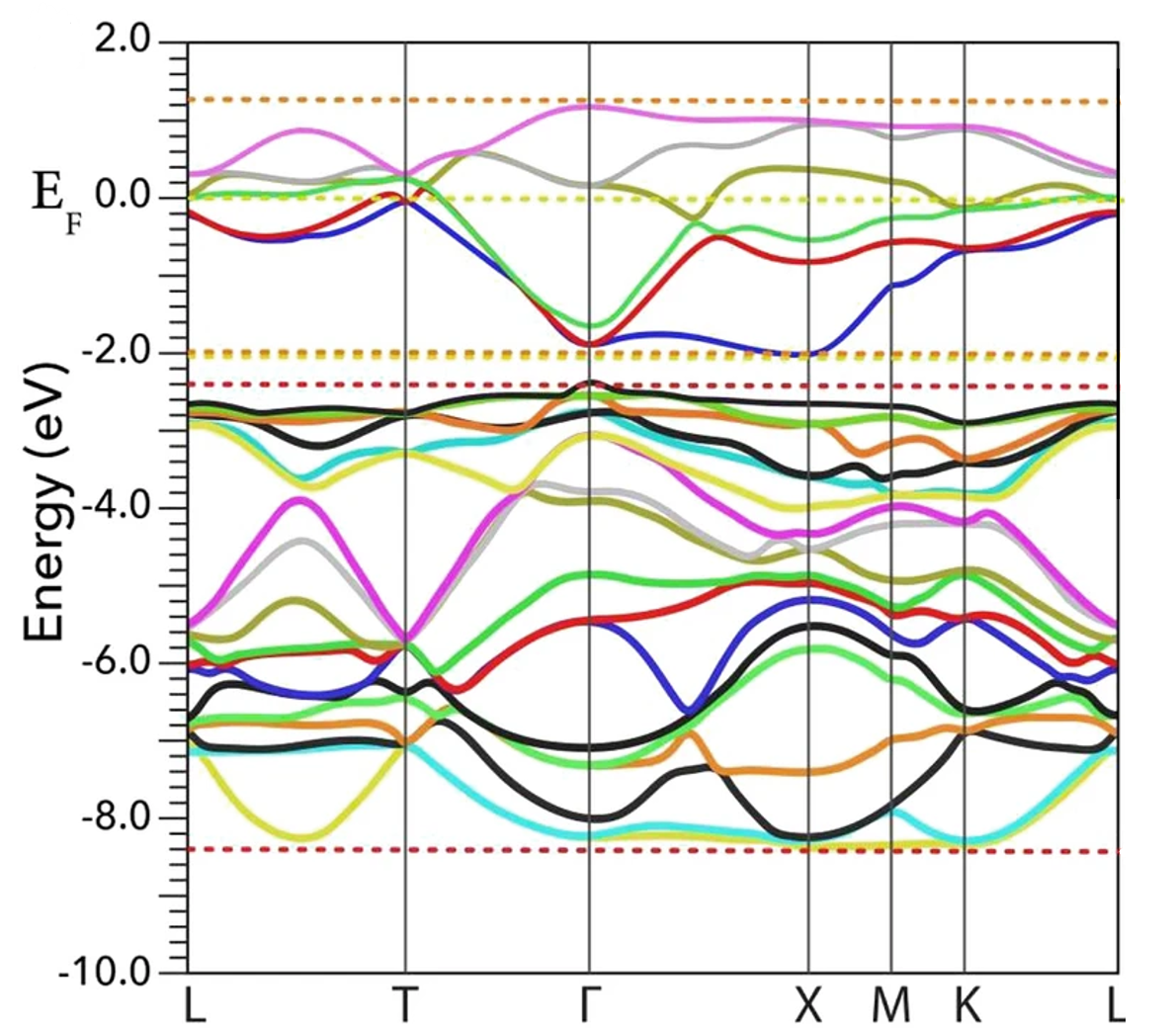
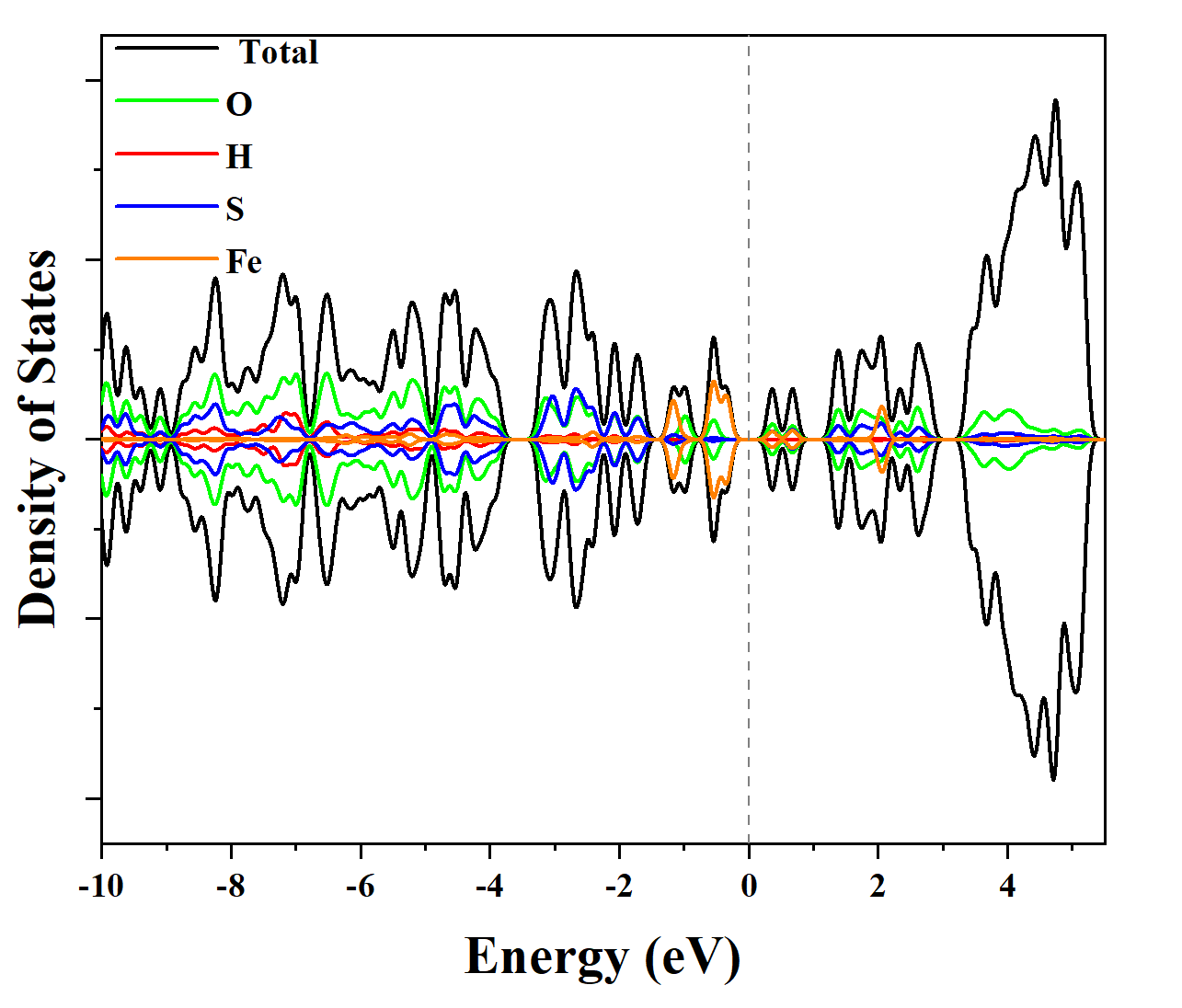
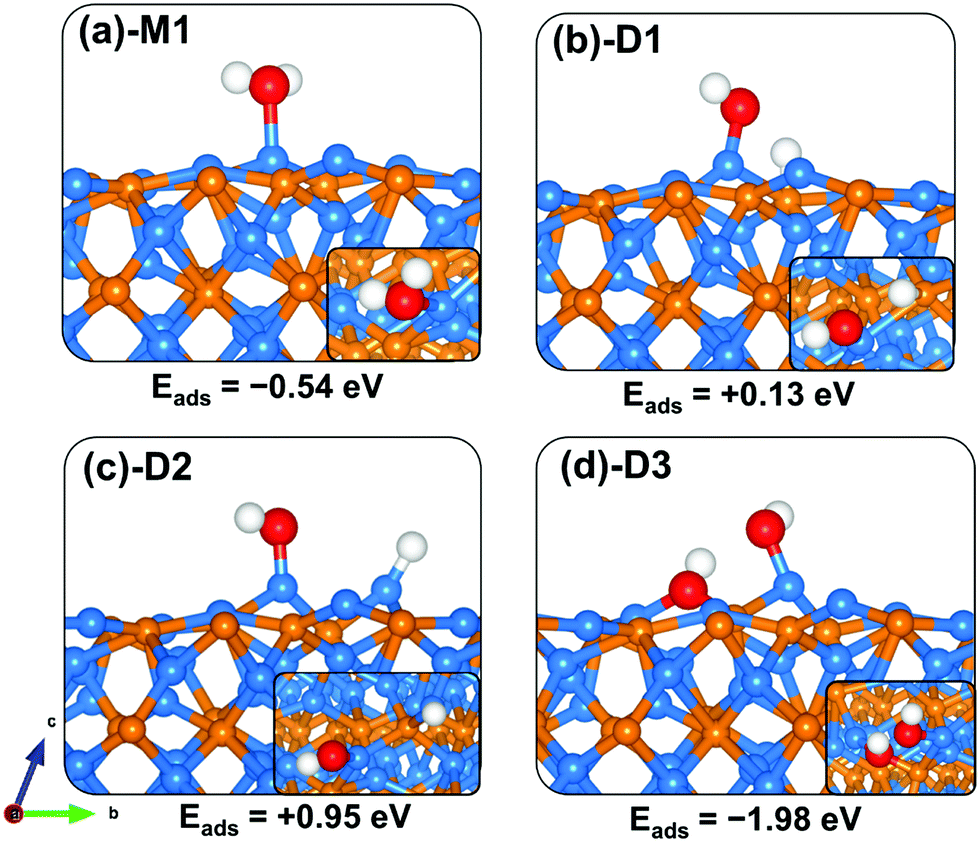
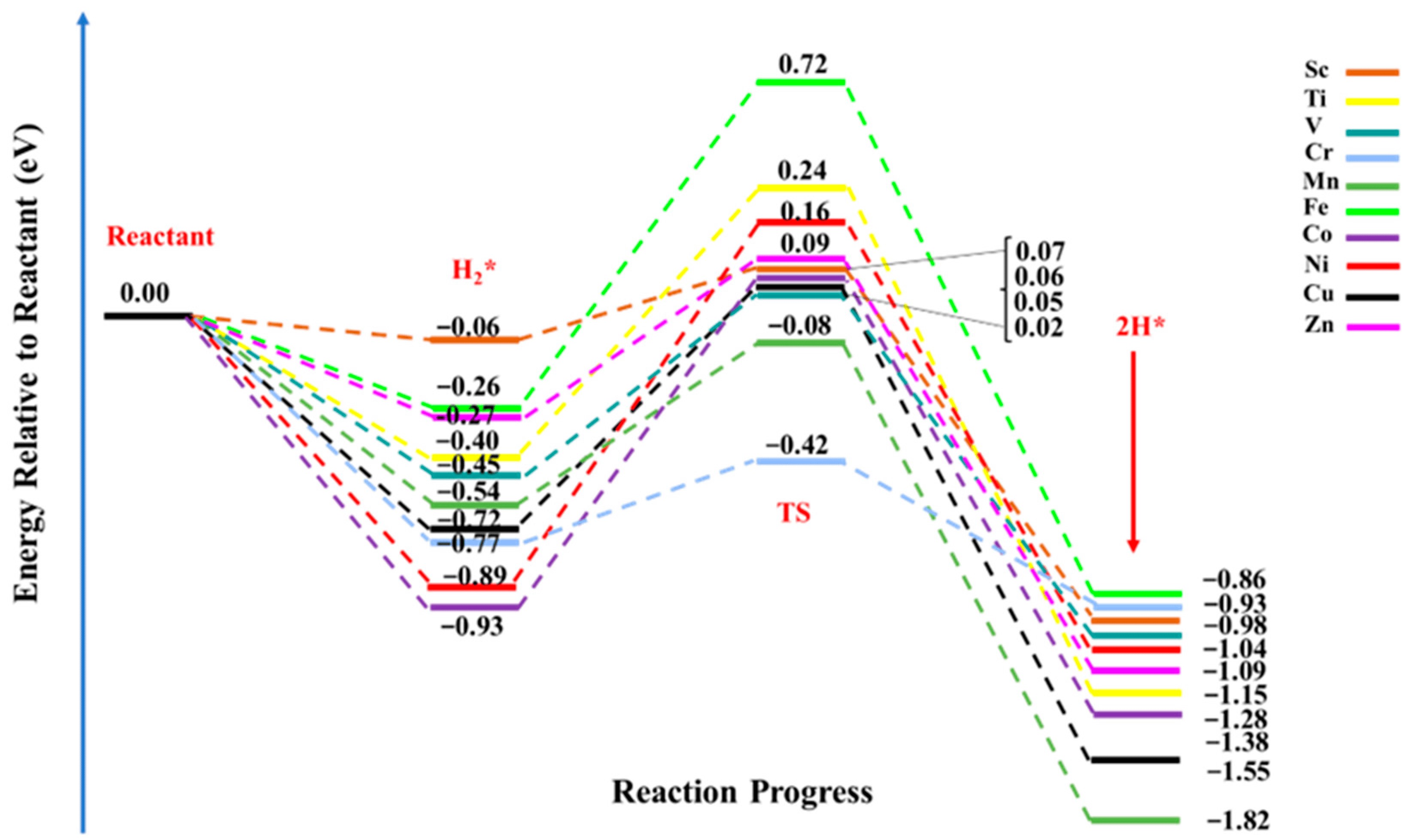
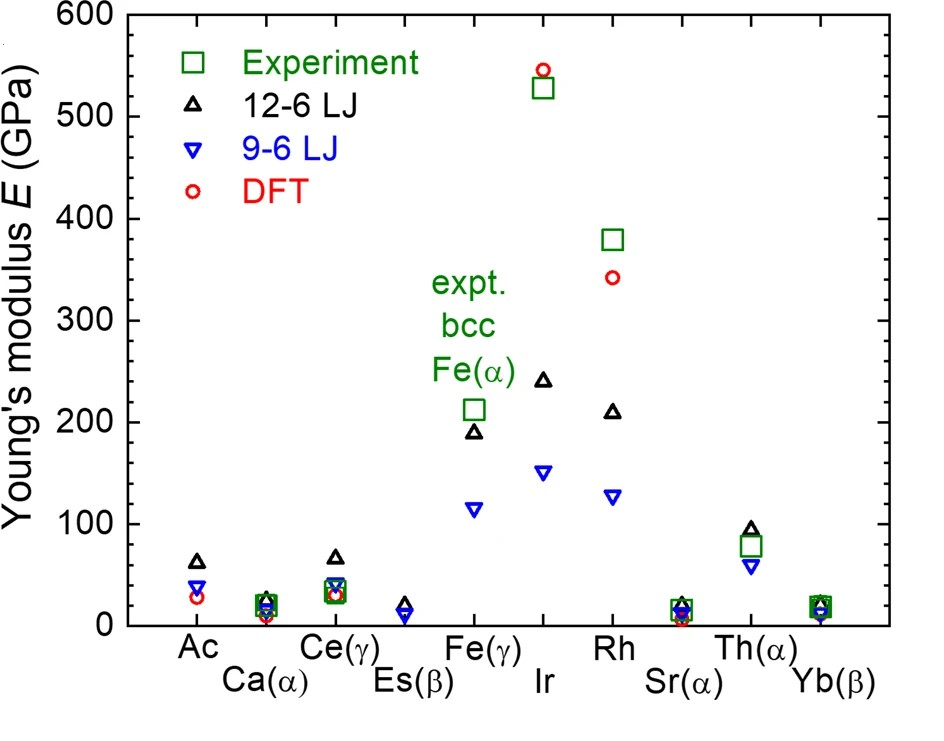
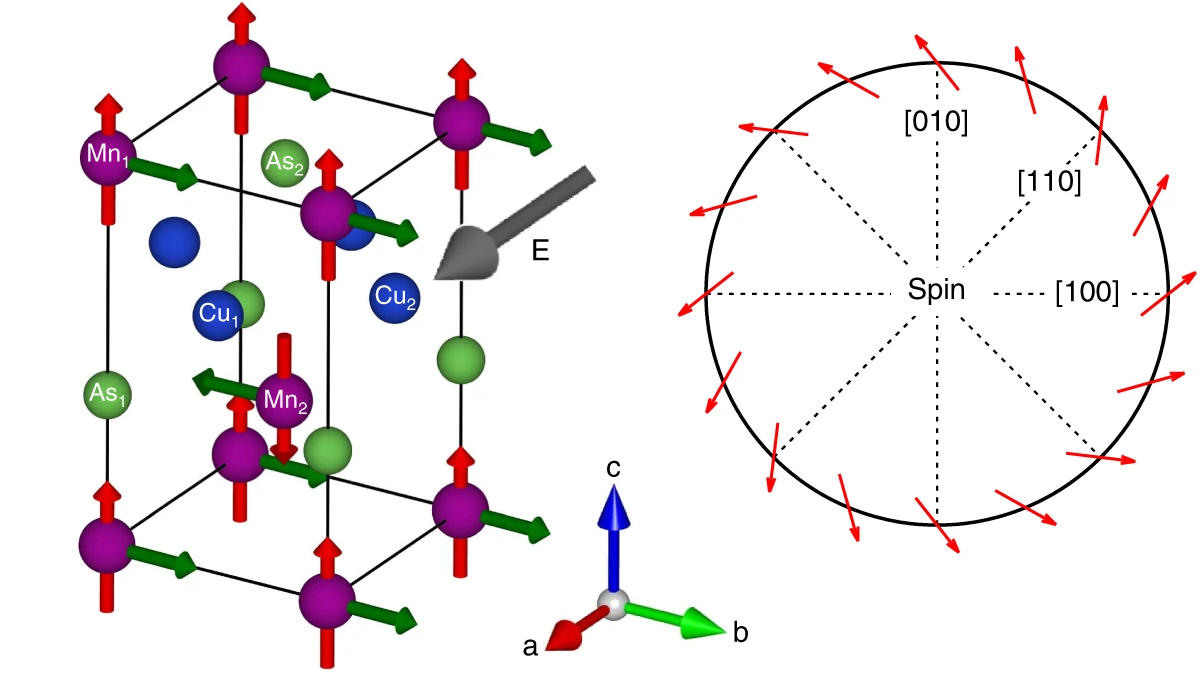
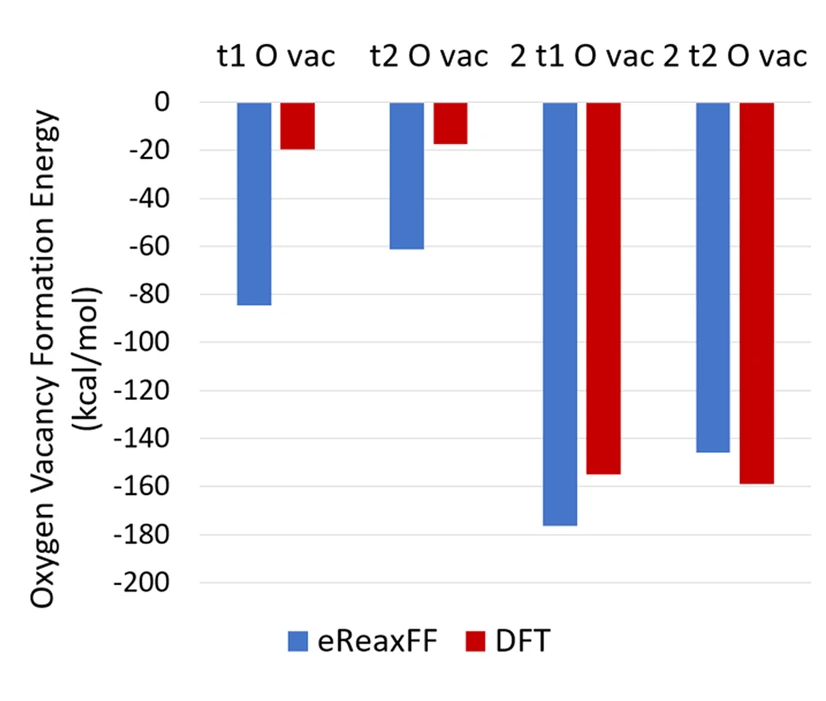
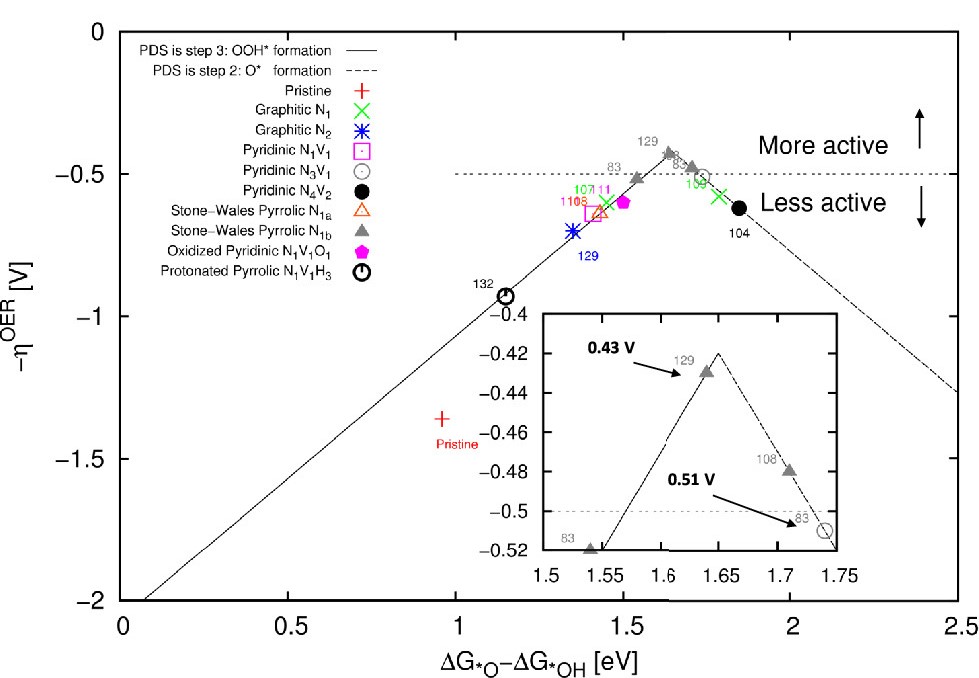
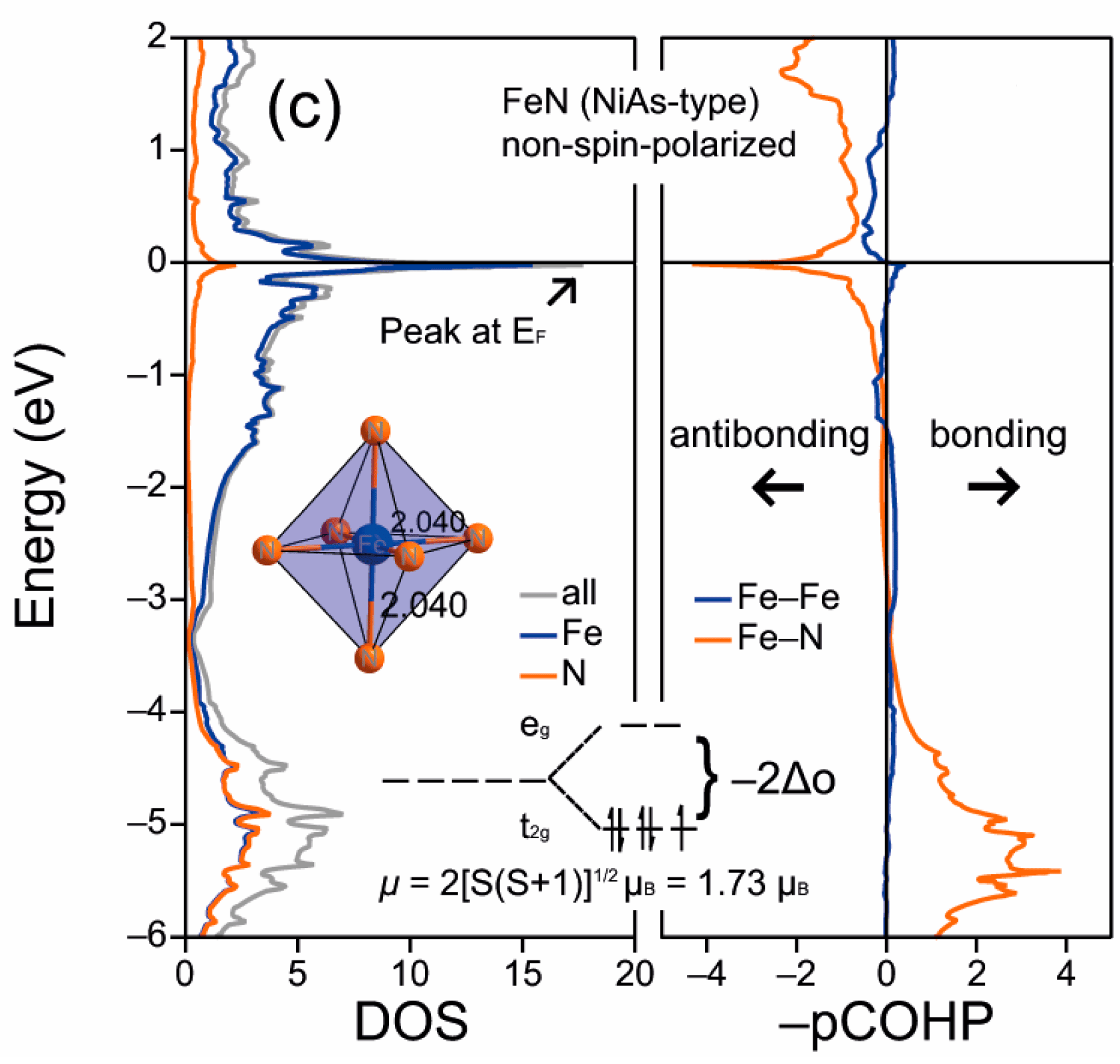
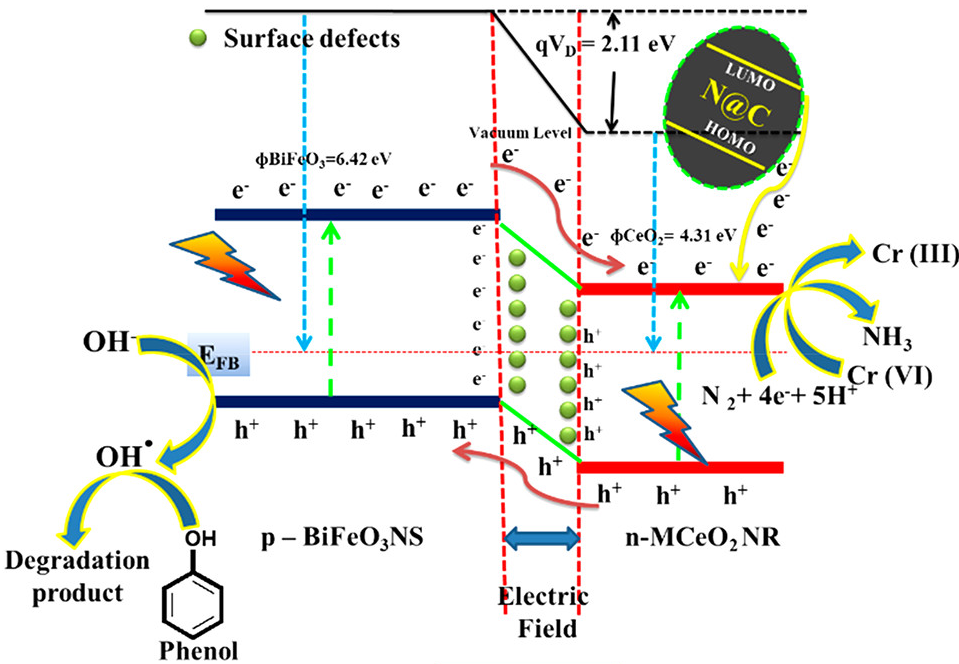


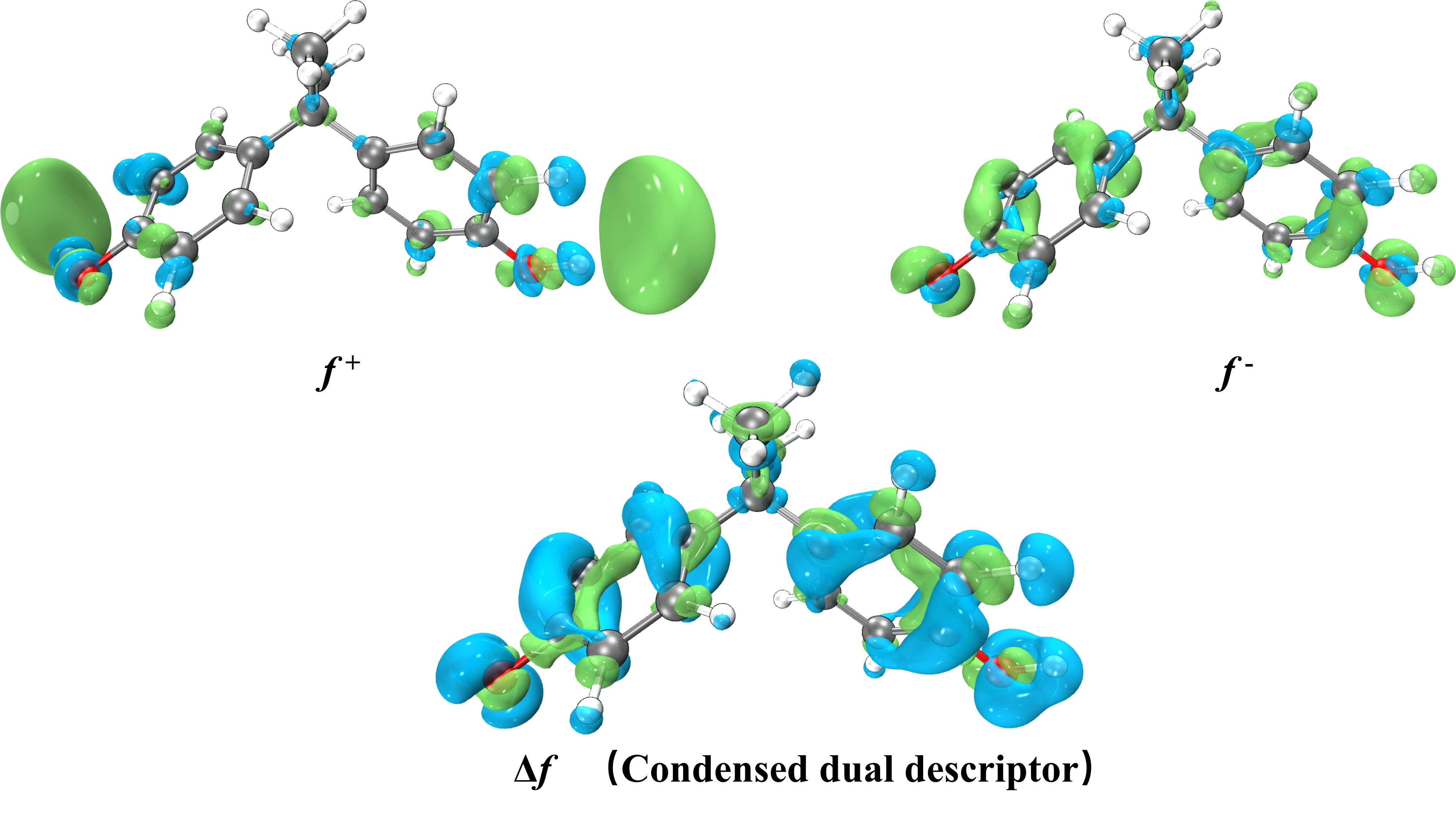
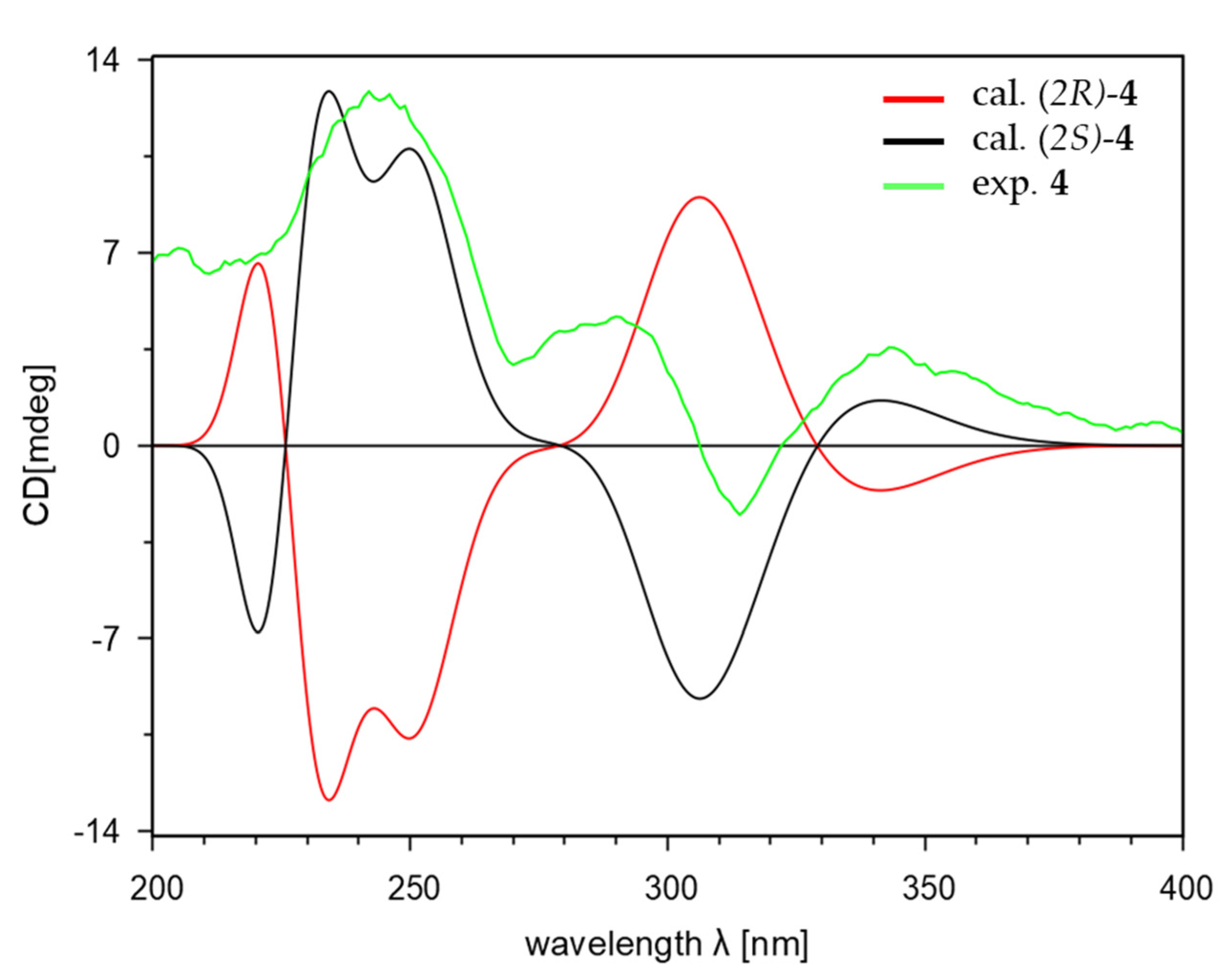
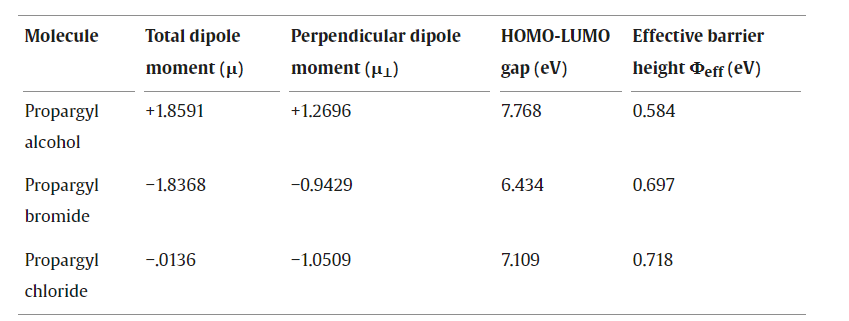
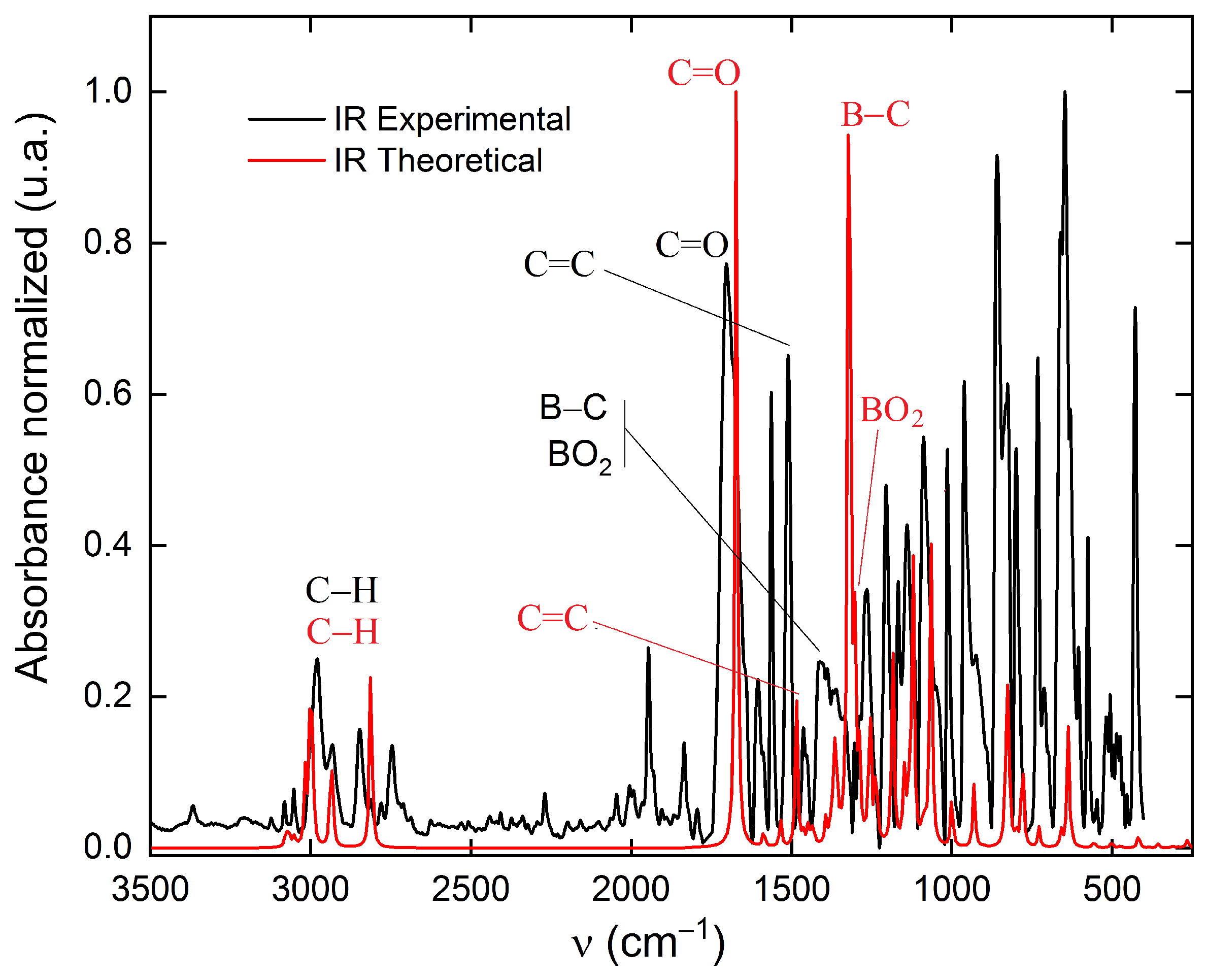
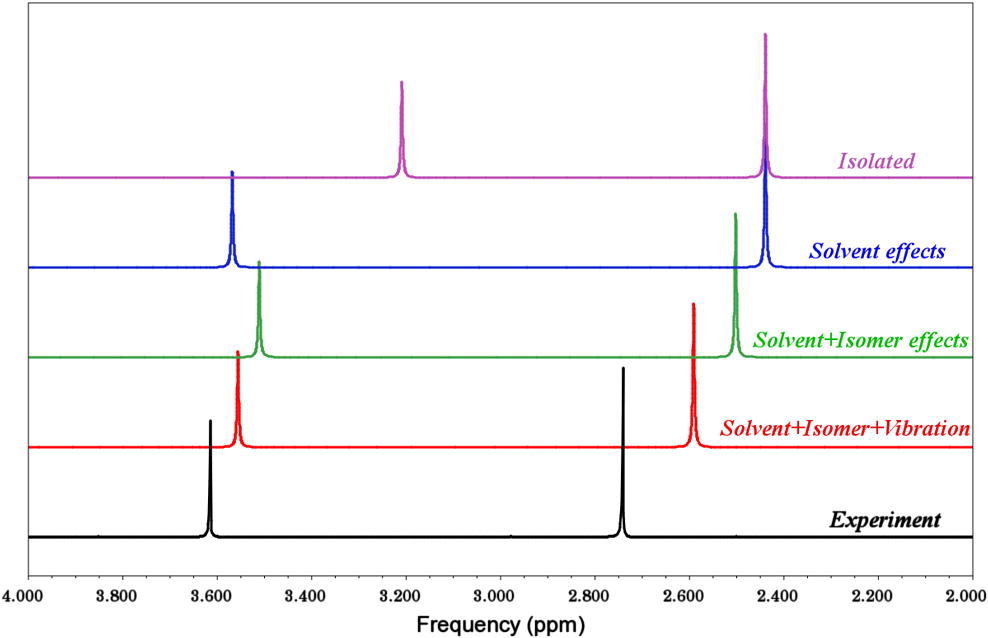
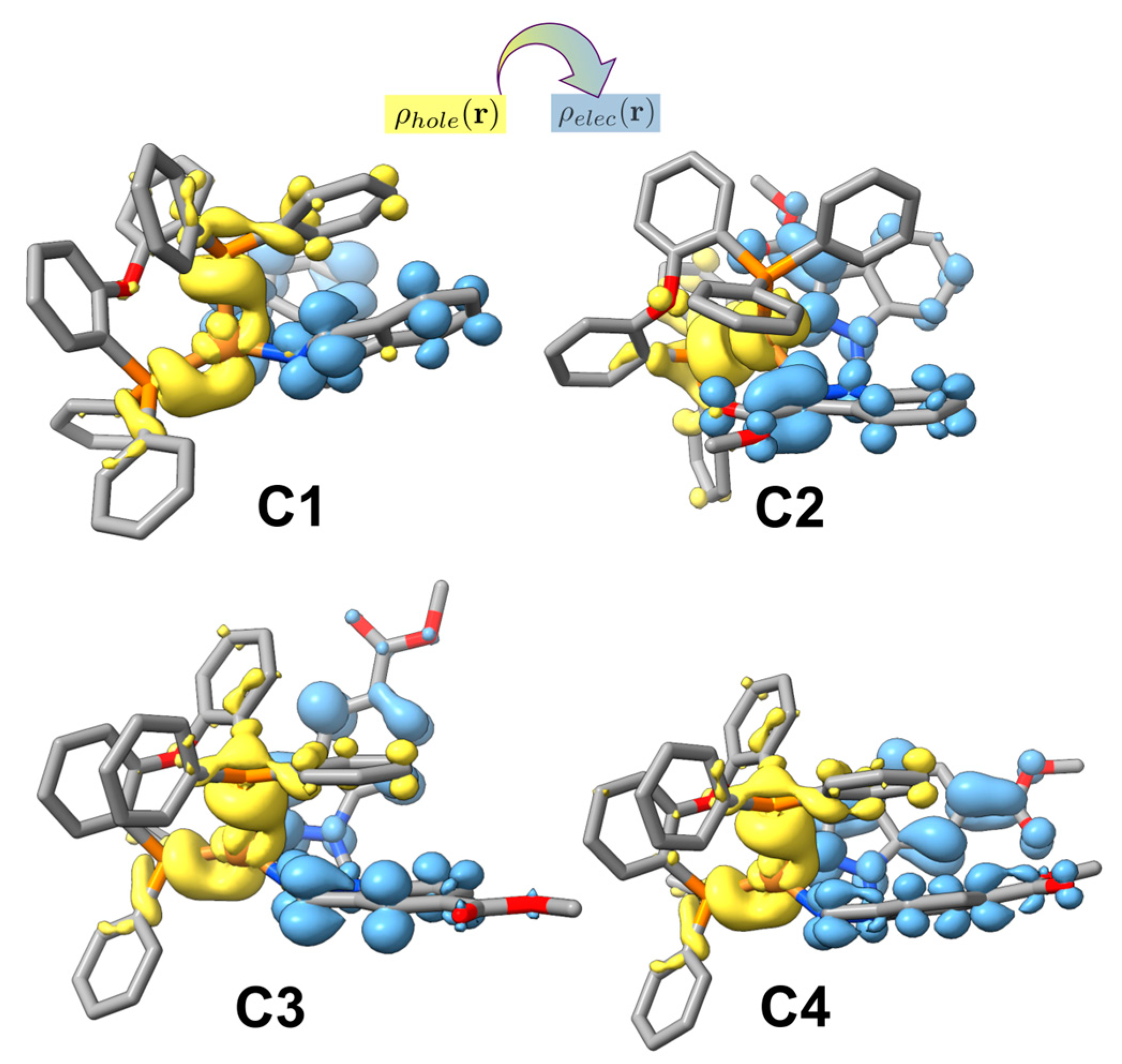
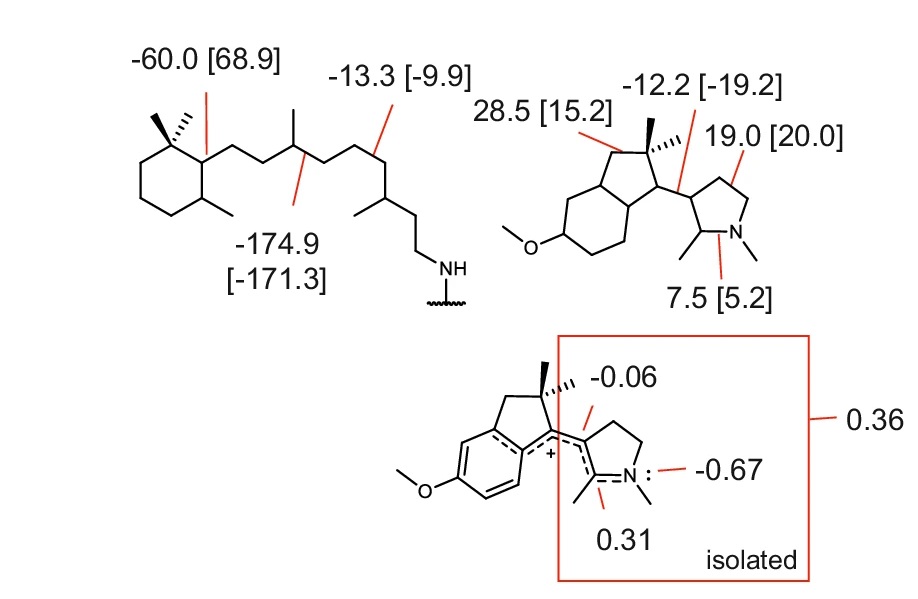
电荷分布、静电势、福井函数、偶极矩、HOMO/ LUMO,电离势、自旋密度、空穴-电子分析、反应机理、反应动力学、过渡态、自由能、结合能、形成焓、激发态、键能、差分电荷密度 、势能面扫描、拉曼光谱、红外光谱、荧光光谱、磷光光谱、圆二色谱、核磁共振谱、旋光度、极化率、振动耦合、成键分析、氢键、卤键、π-π堆积、硫键、疏水作用力、Hirshfeld表面分析、独立梯度模型、构象搜索

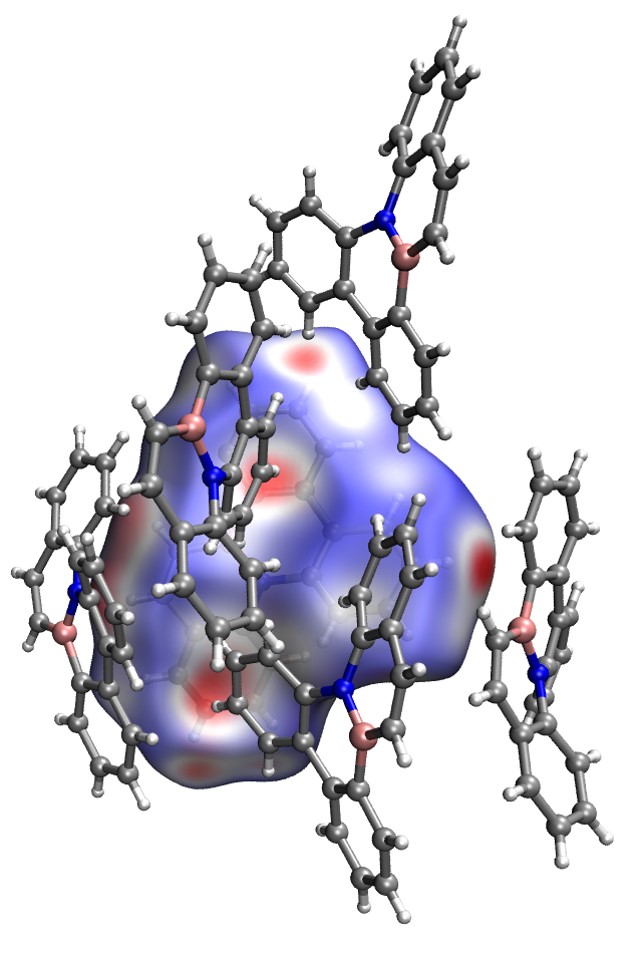
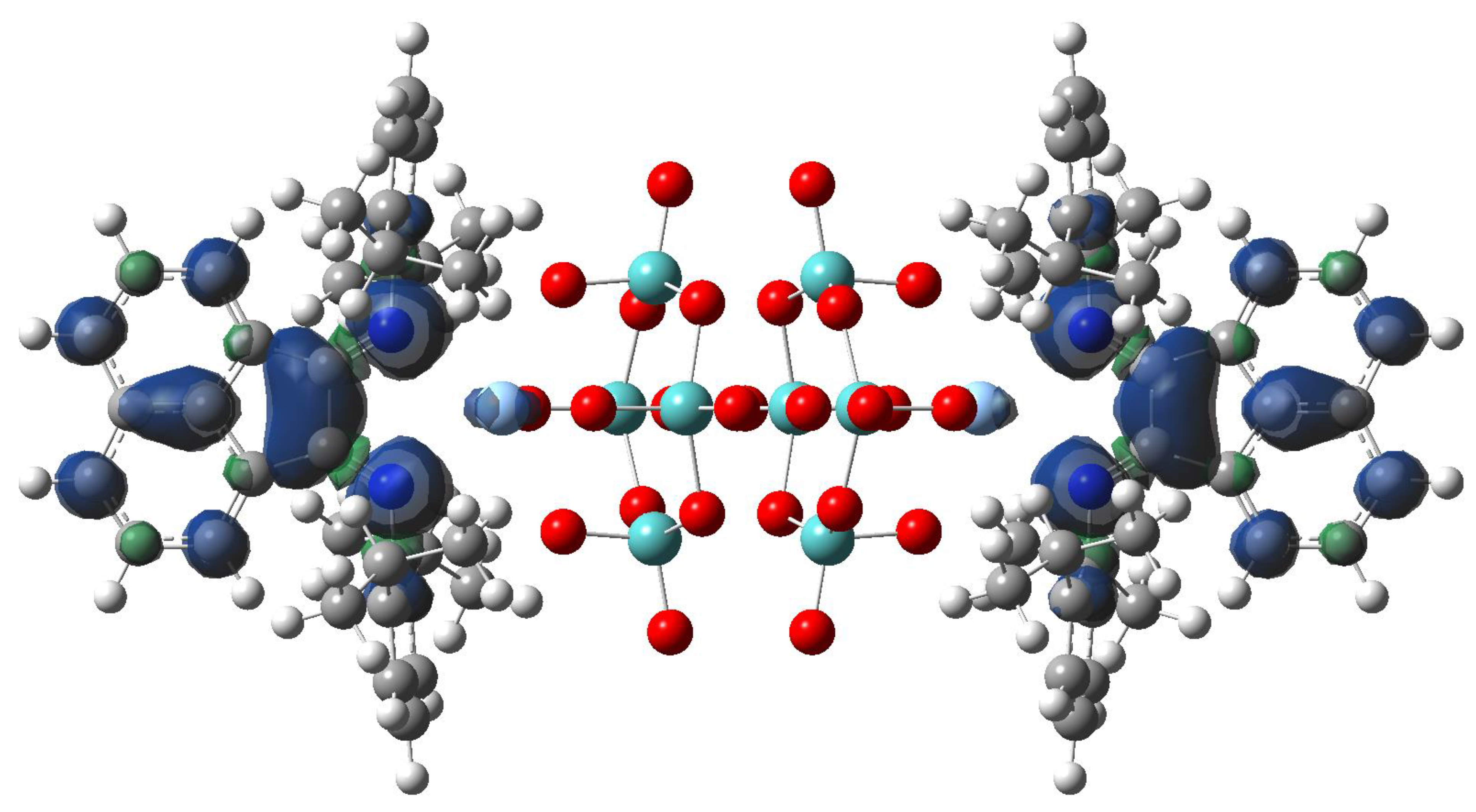

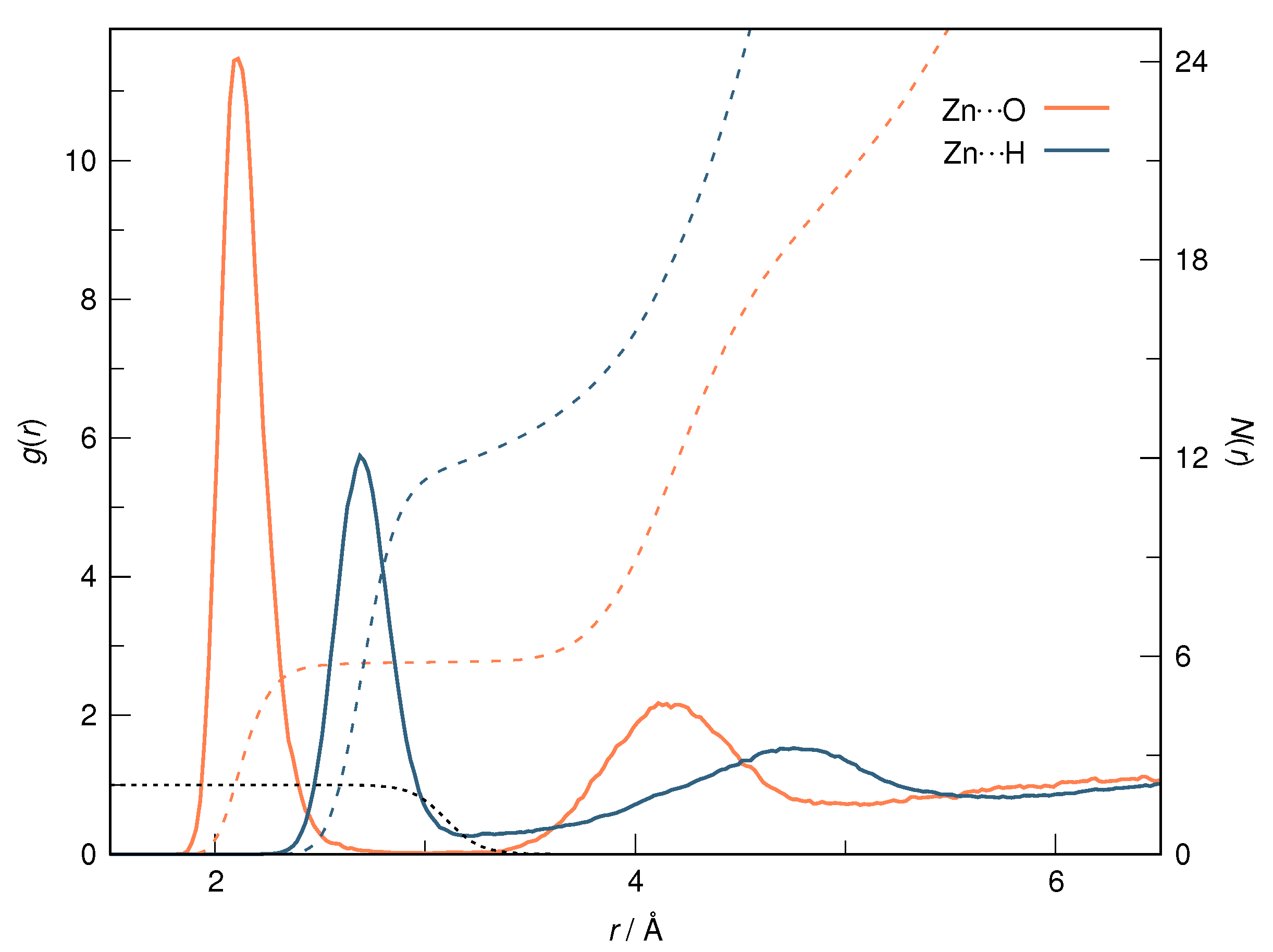
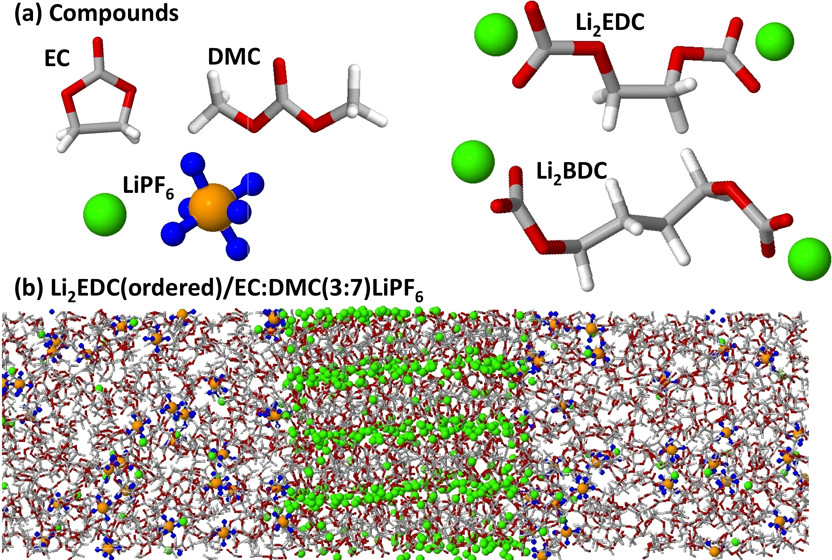
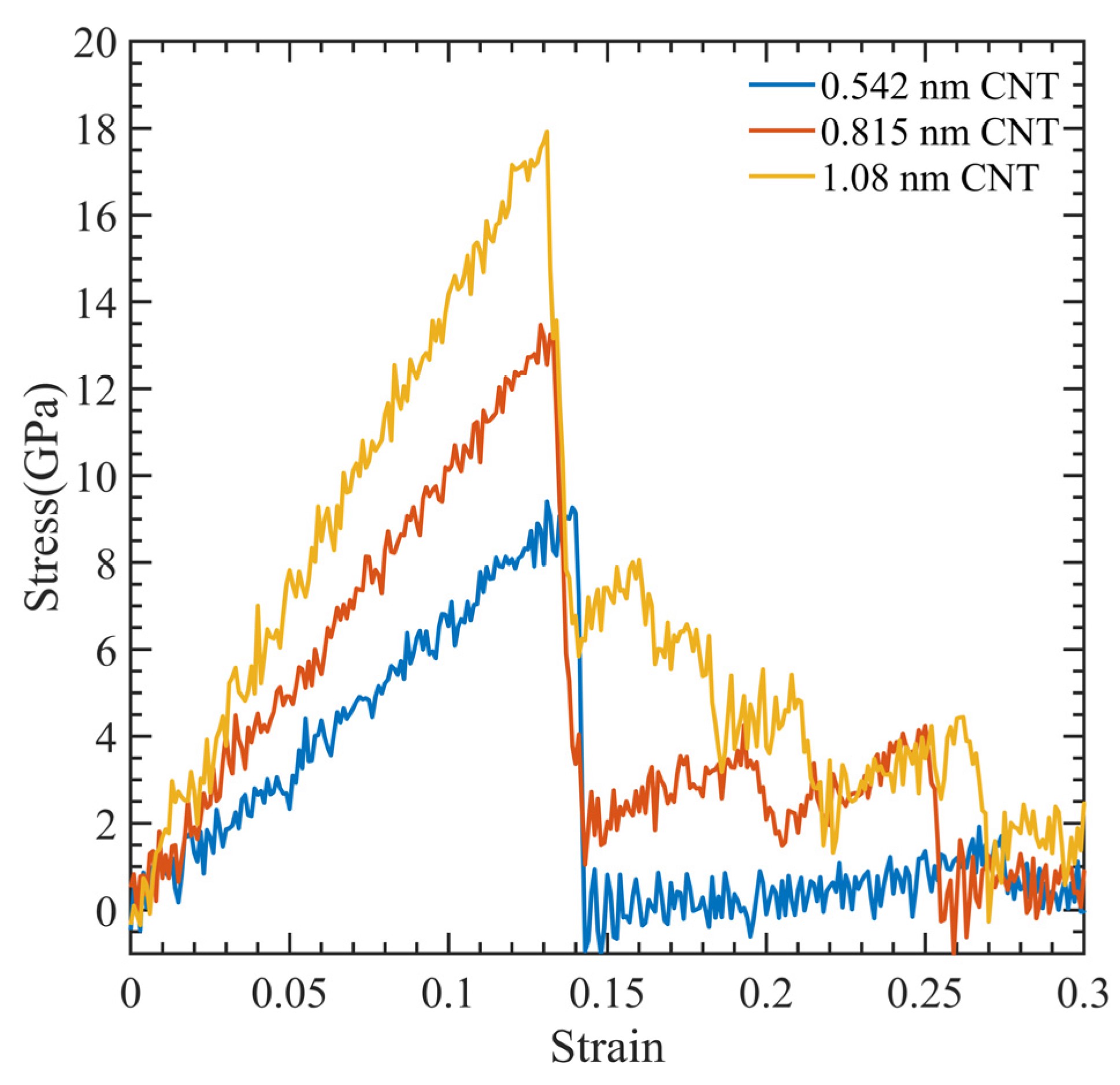
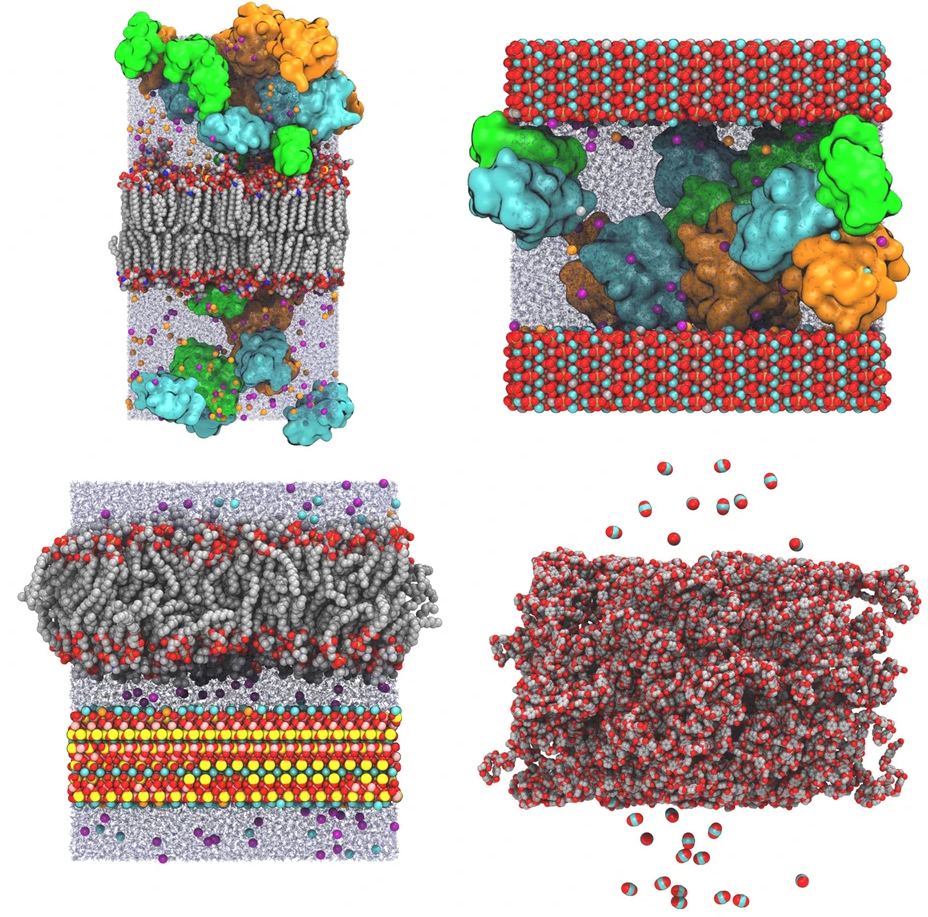
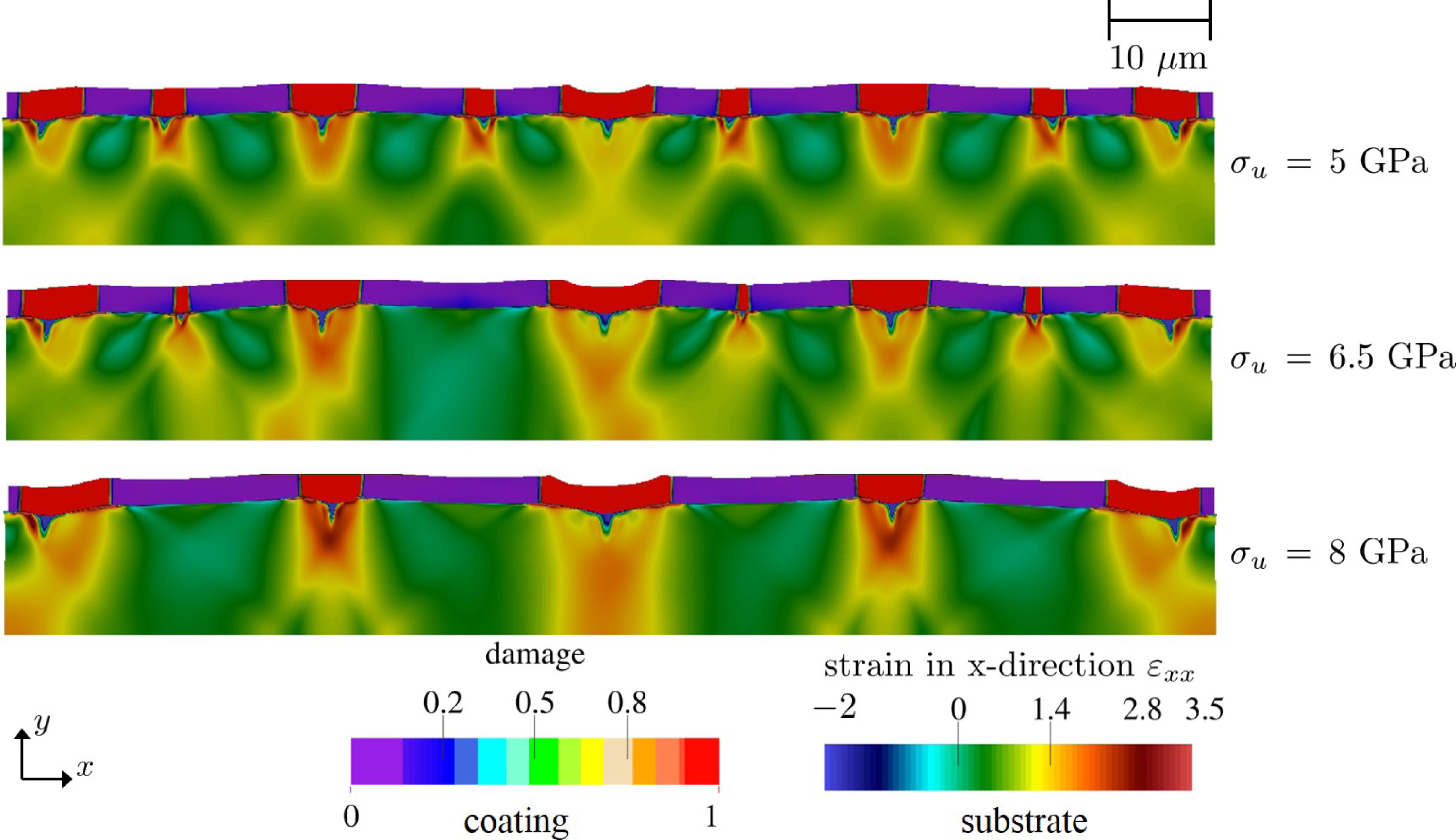
径向分布函数(RDF)、均方位移(MSD)、平均力势(PMF)、扩算系数、密度分布、氢键分析、相互作用力、自由能形貌图、界面润湿、界面扩散、生物大分子动力学模拟、电池电解液、溶剂化结构、分子相互作用、非晶结构、玻璃化转变温度、压痕、切削、摩擦、热导率、燃烧、热解、焊接、自组装、合金热稳定性、熔点、热膨胀率、相转变、结晶、晶格位错、应力应变曲线、自由体积、化学抛光




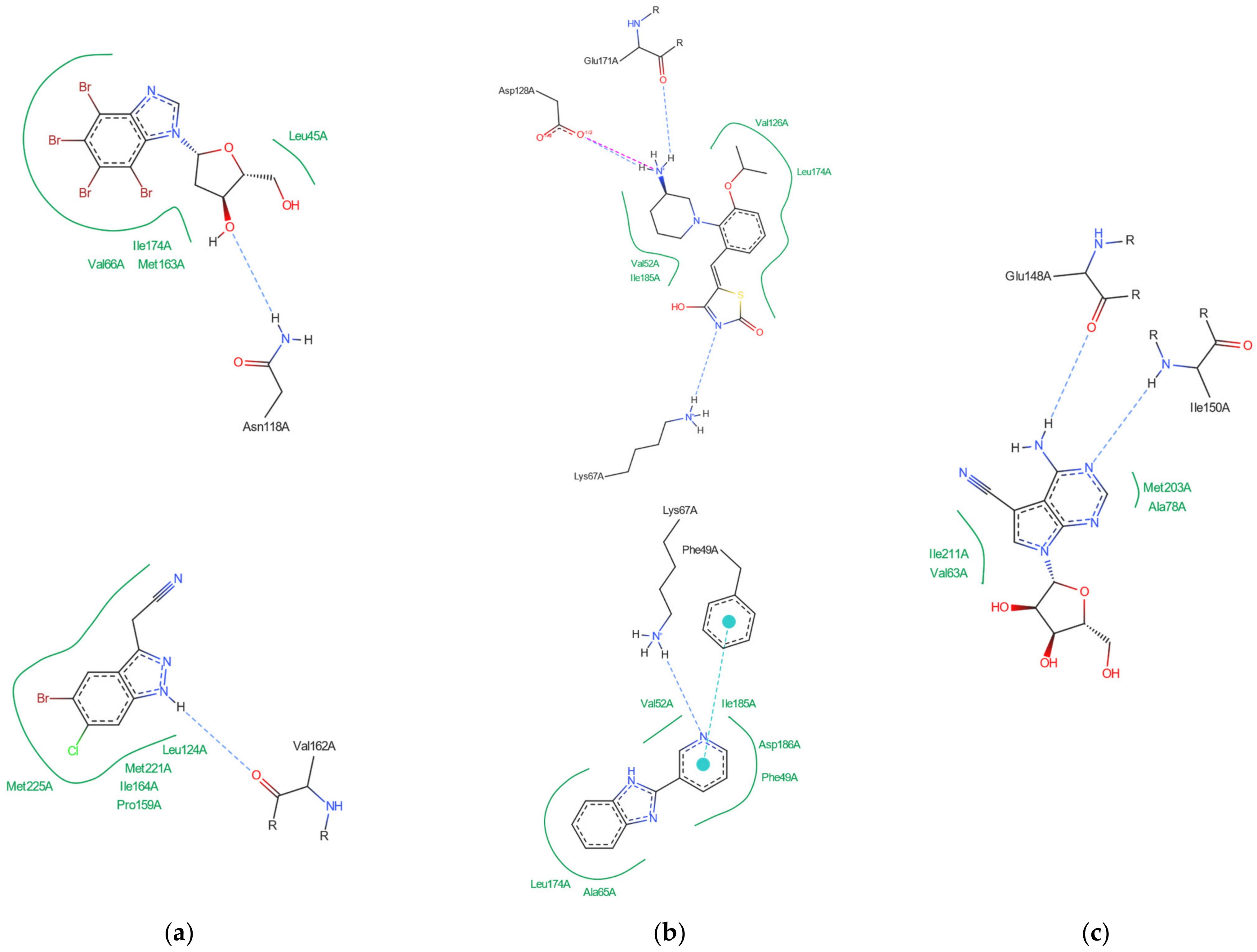
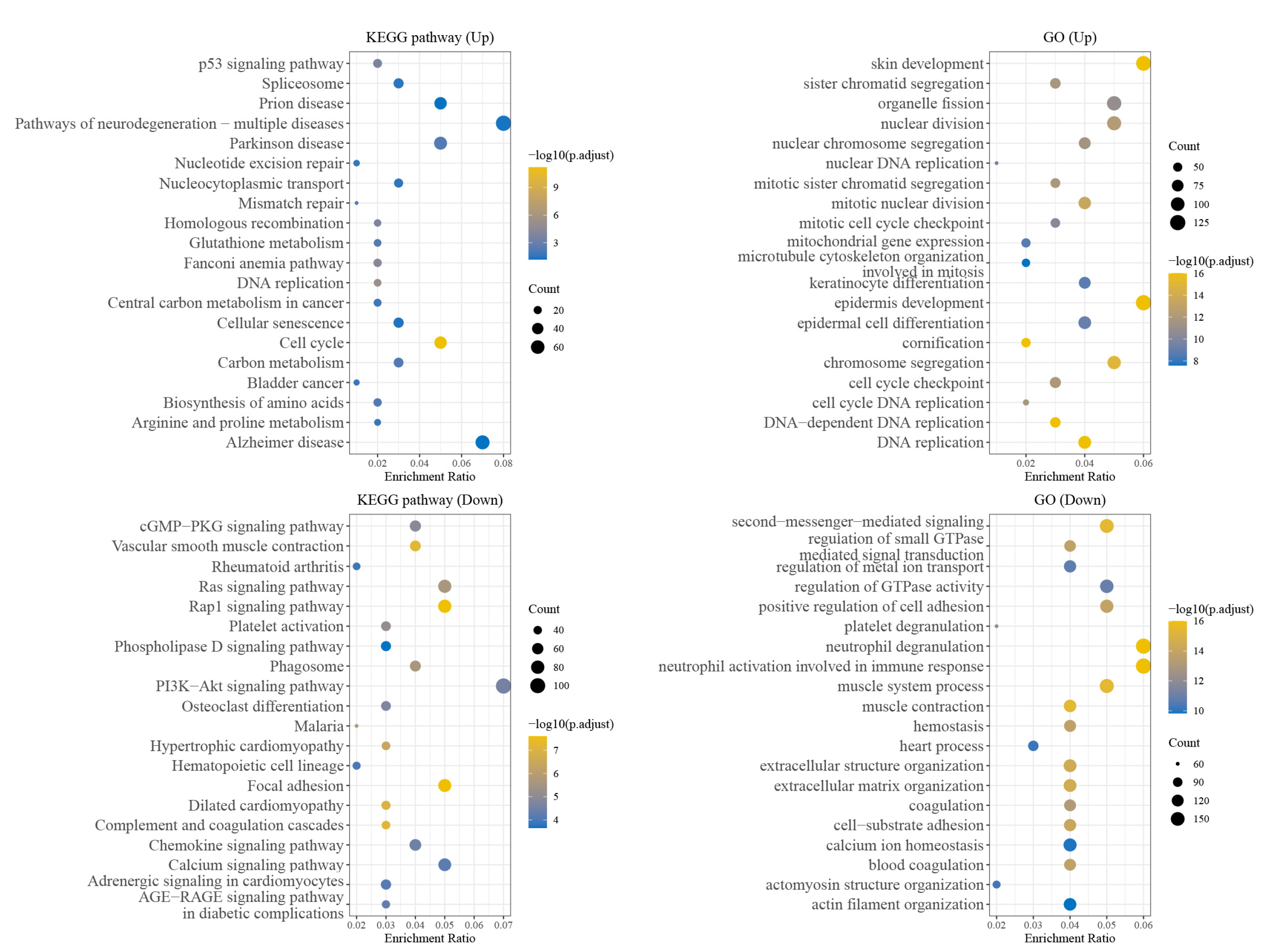
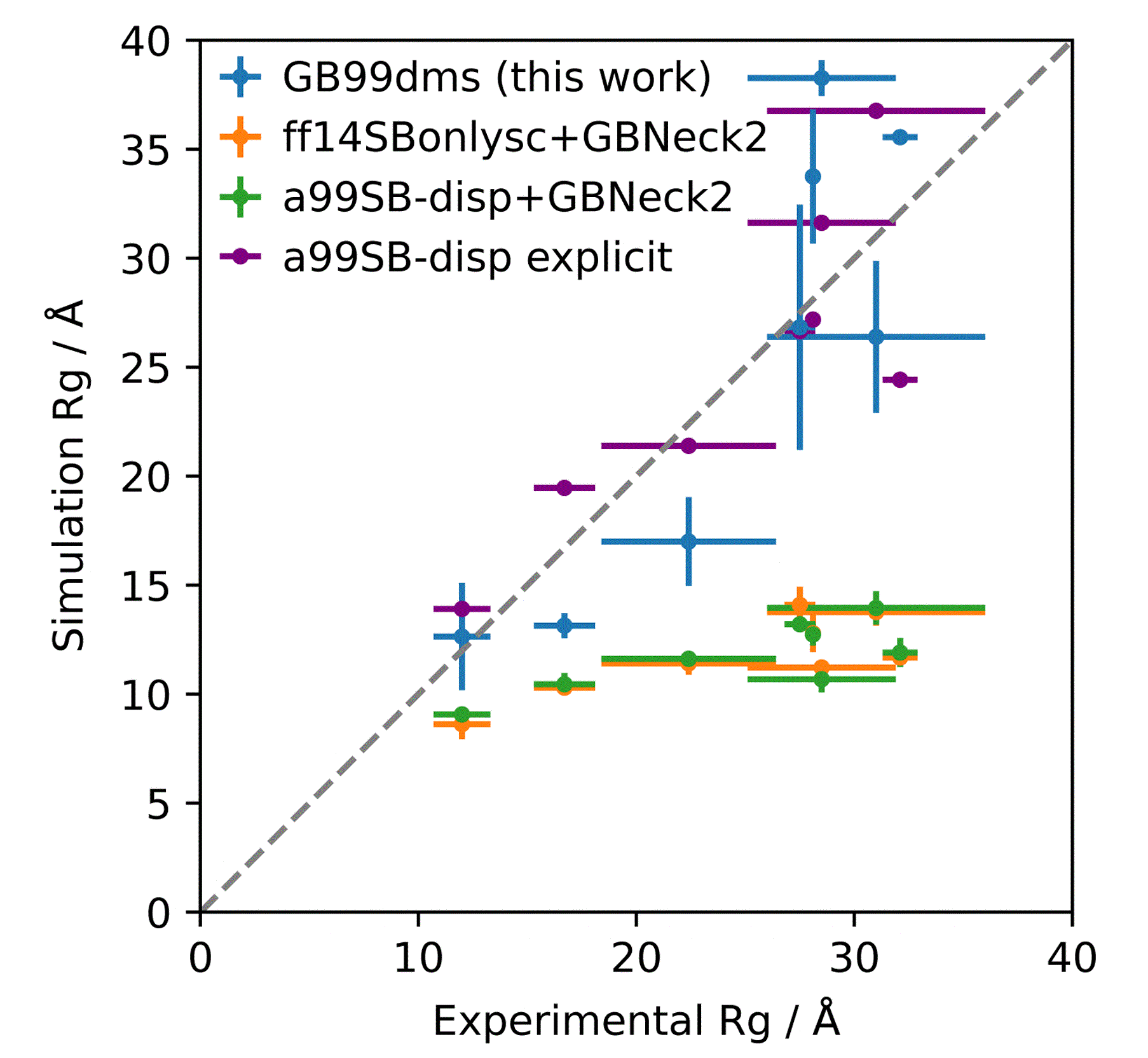
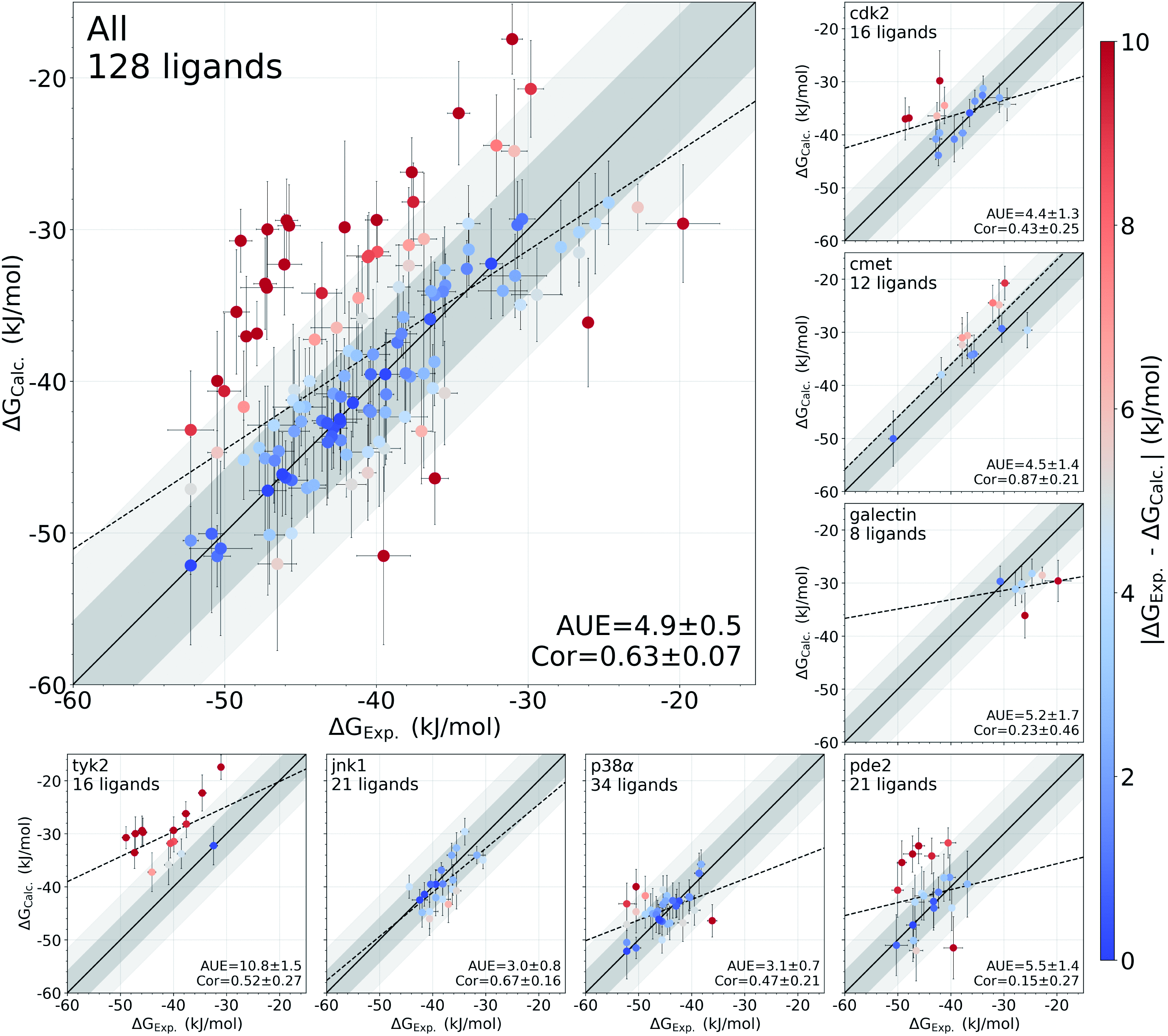
分子对接、虚拟筛选、同源建模、成药性预测、配体-受体结合、病毒研究、多肽设计、生信分析、酶催化、结合自由能、QM/MM、粗粒化模拟、自组装、蛋白结构预测、蛋白拉伸、分子间相互作用、能量分析、成键分析、氢键分析、结构聚类分析 、分子动力学、回旋半径(Rg)、均方根偏差(RMSD)、均方根涨落(RMSF)
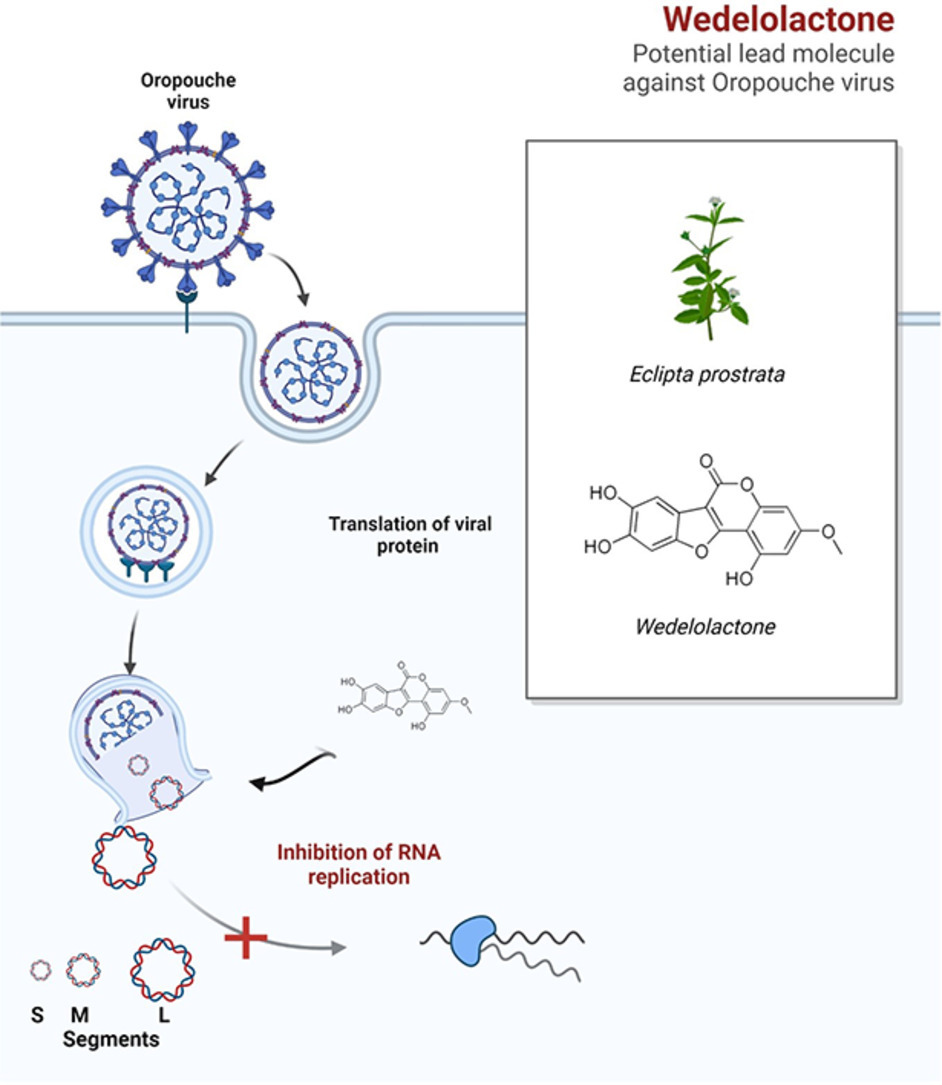
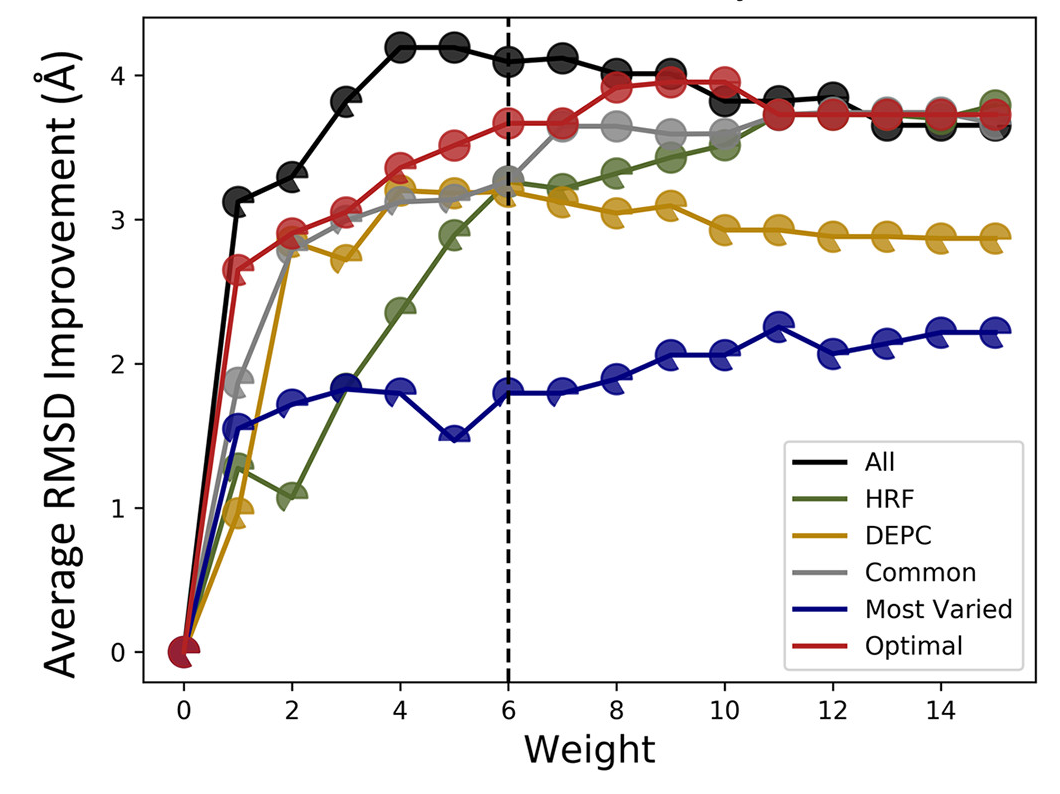
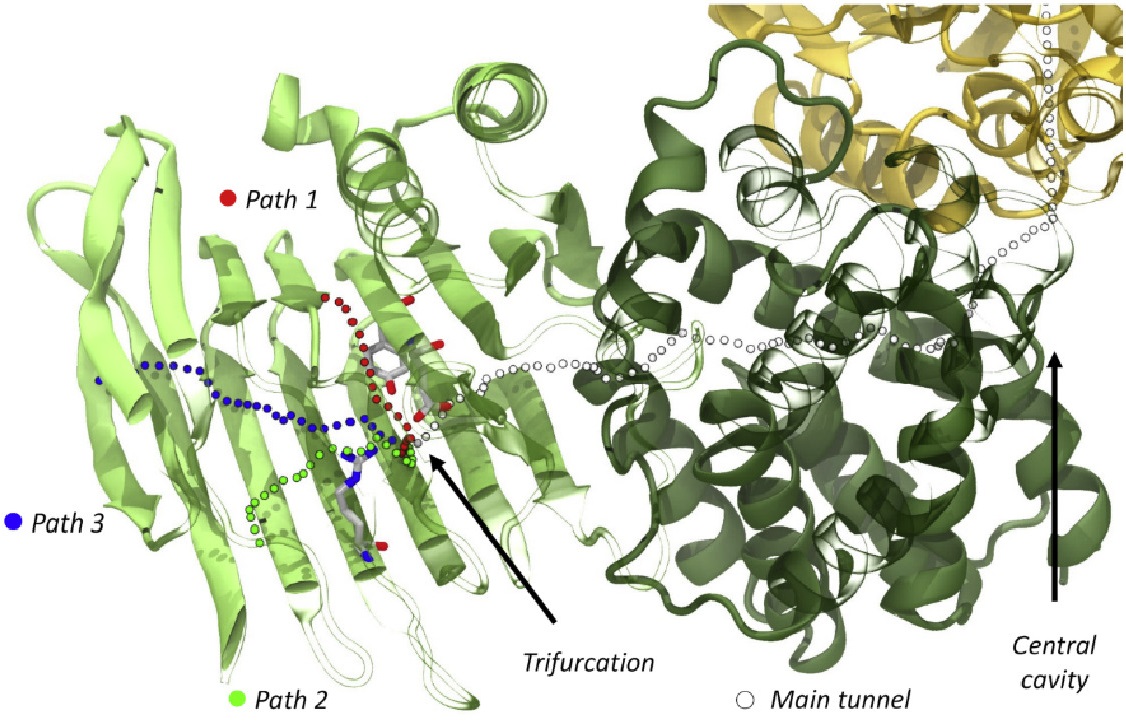
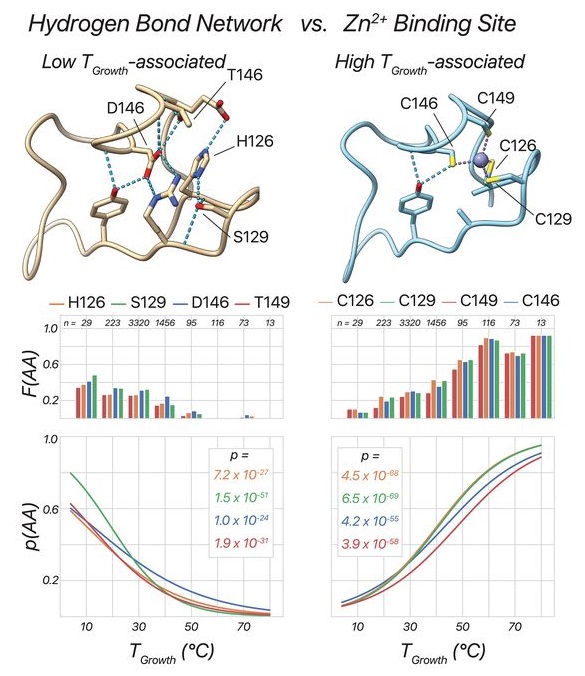
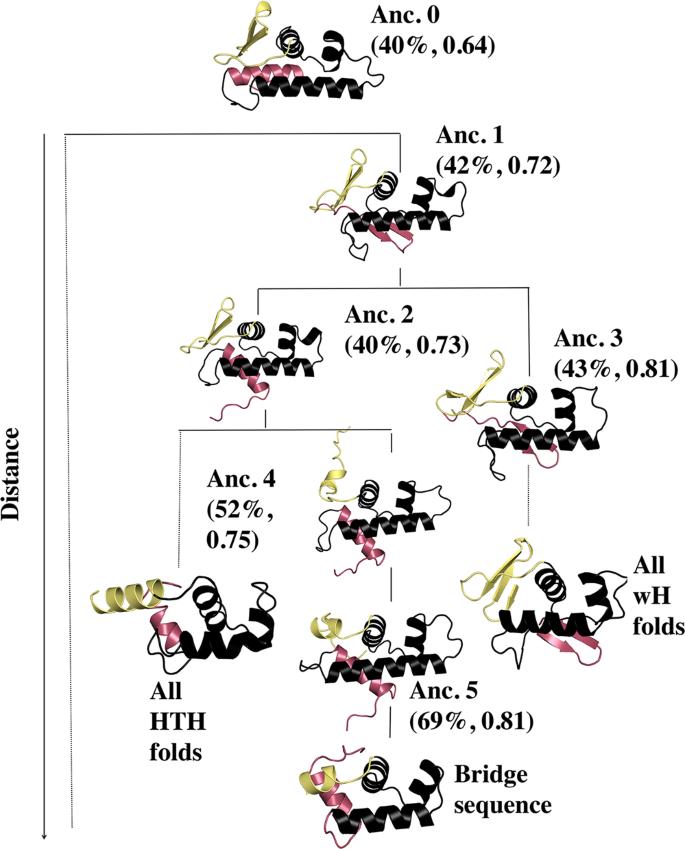
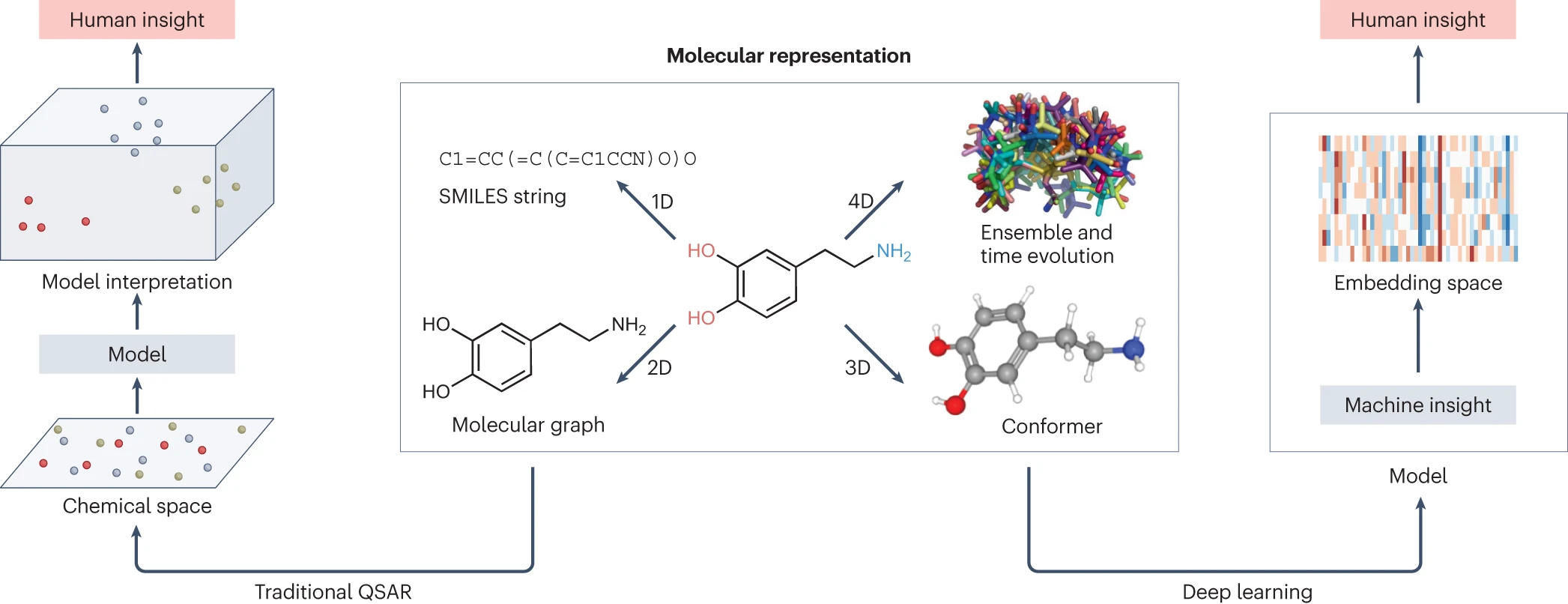
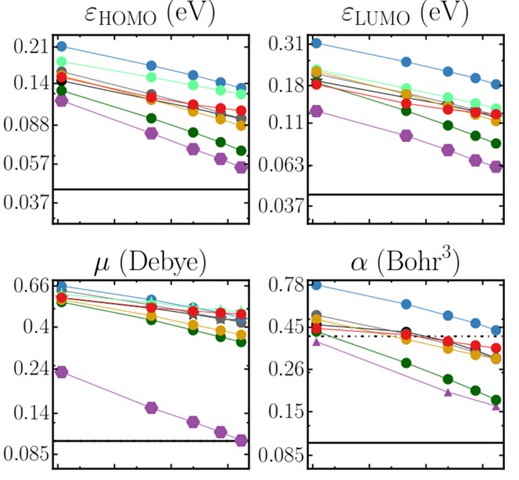
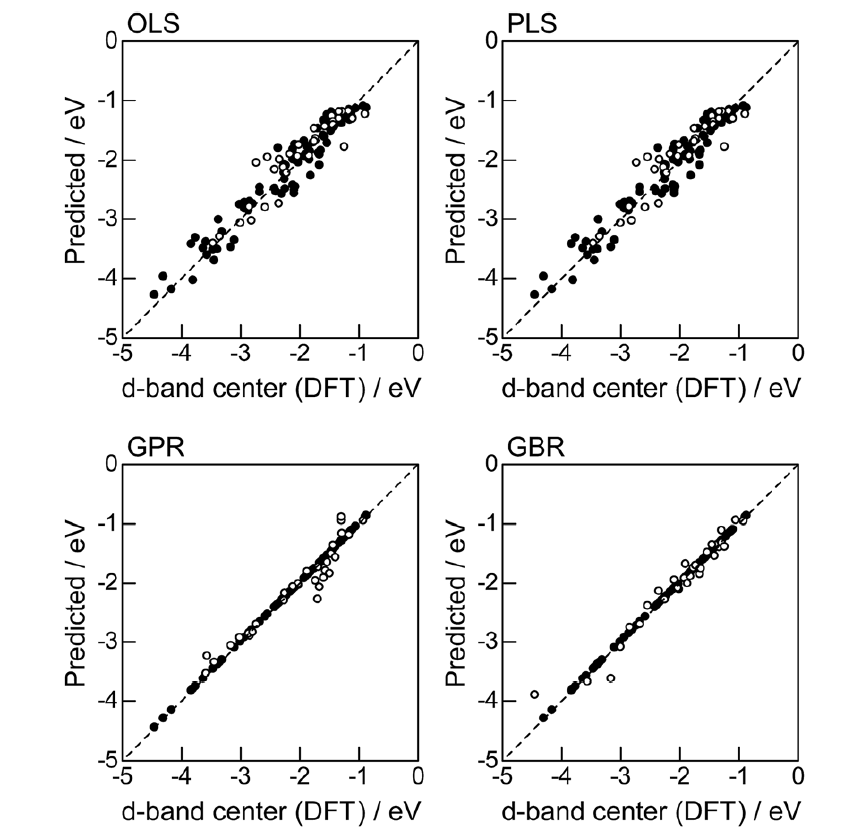
高通量筛选、势函数训练、预测材料的催化性能、预测反应路径/机理、预测吸附能、预测d带中心、预测结构性质(高熵合金、钙钛矿、二维材料、电解液、膜电极等)、神经网络、线性回归、KNN、决策树、随机森林、支持向量机、聚类算法、模型预测、药物设计、数据分析与挖掘、数据可视化、数据库构建和管理、数据检索/下载
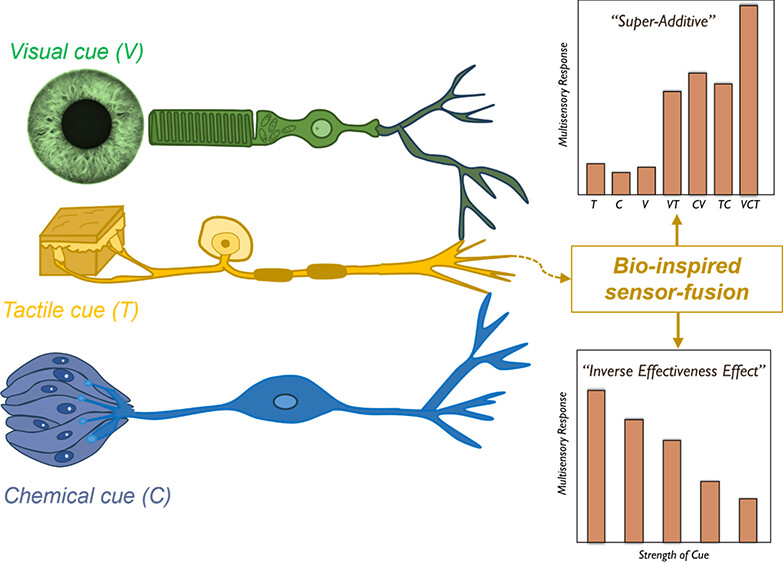
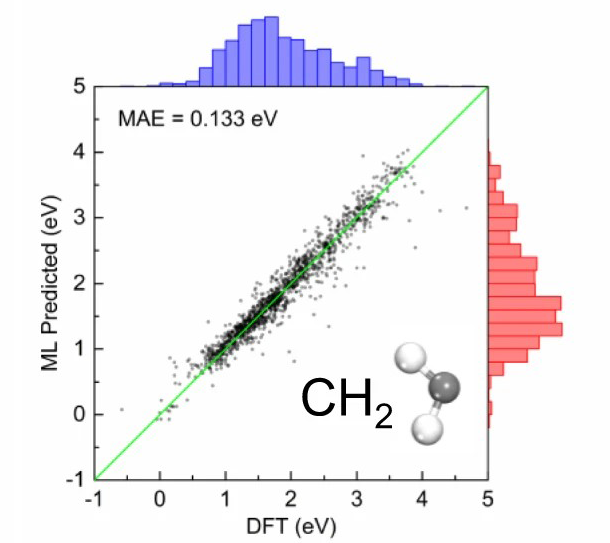
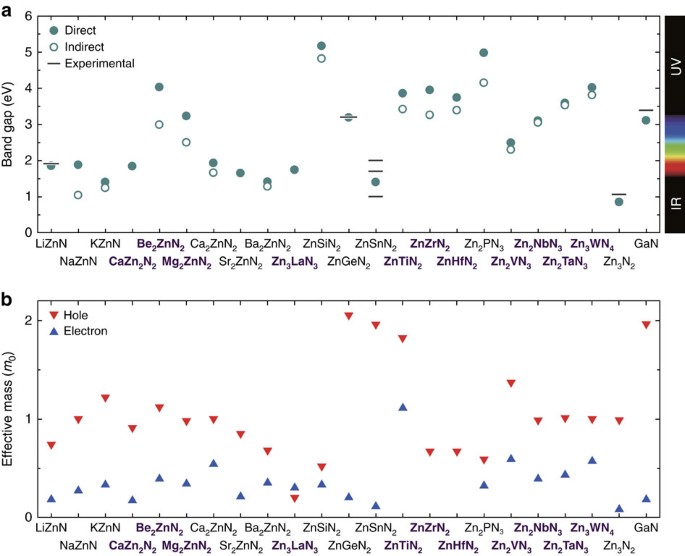
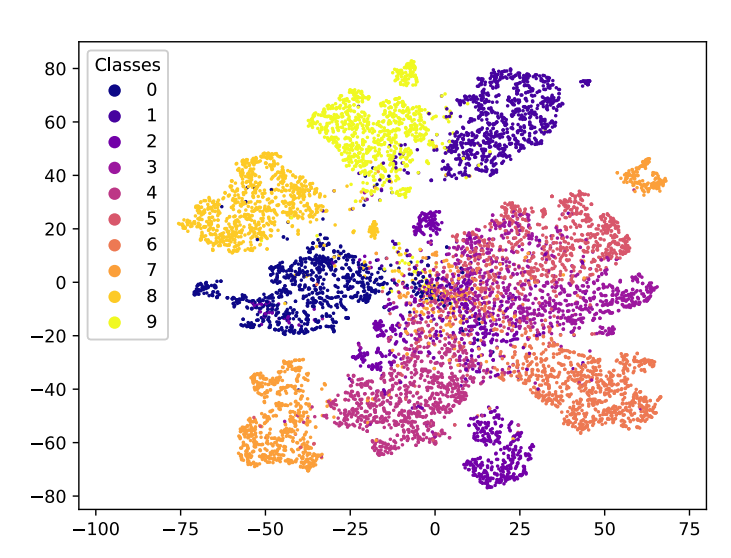
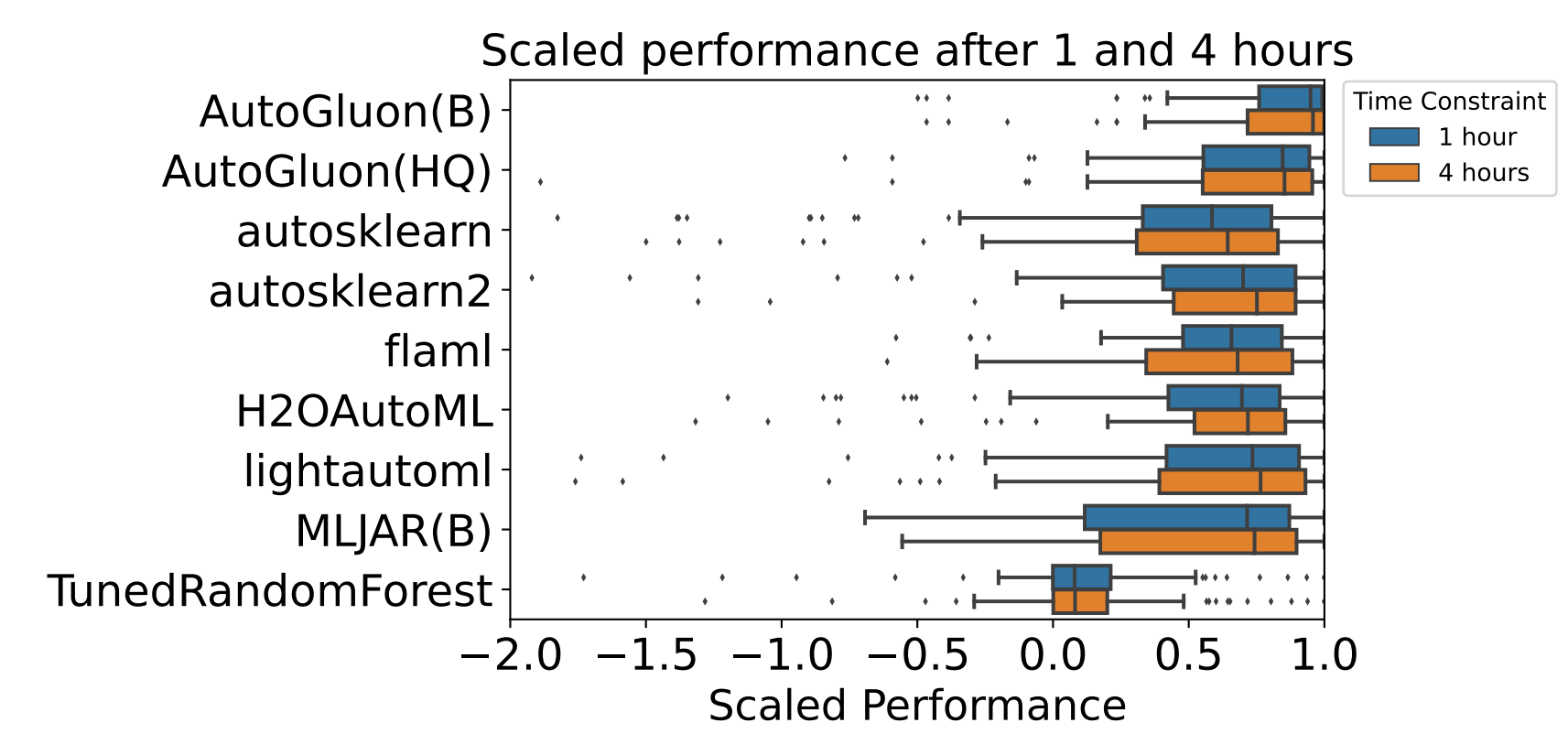

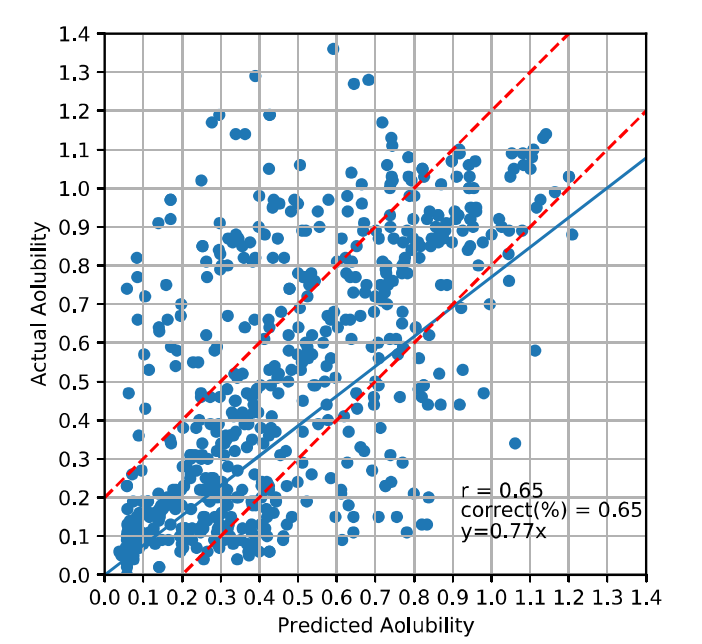
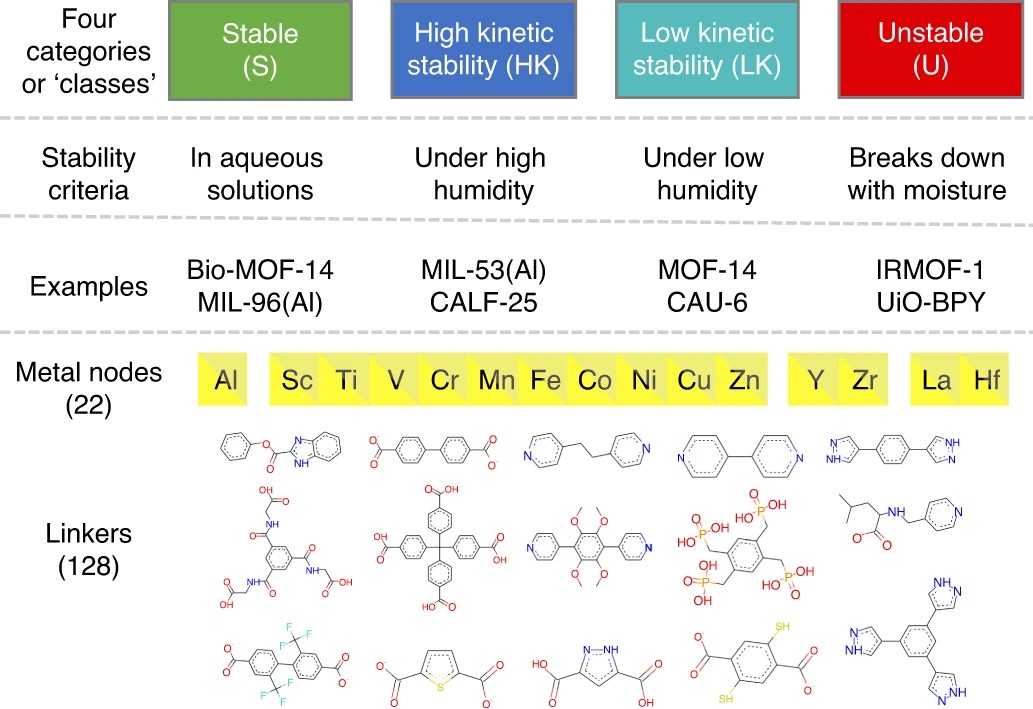
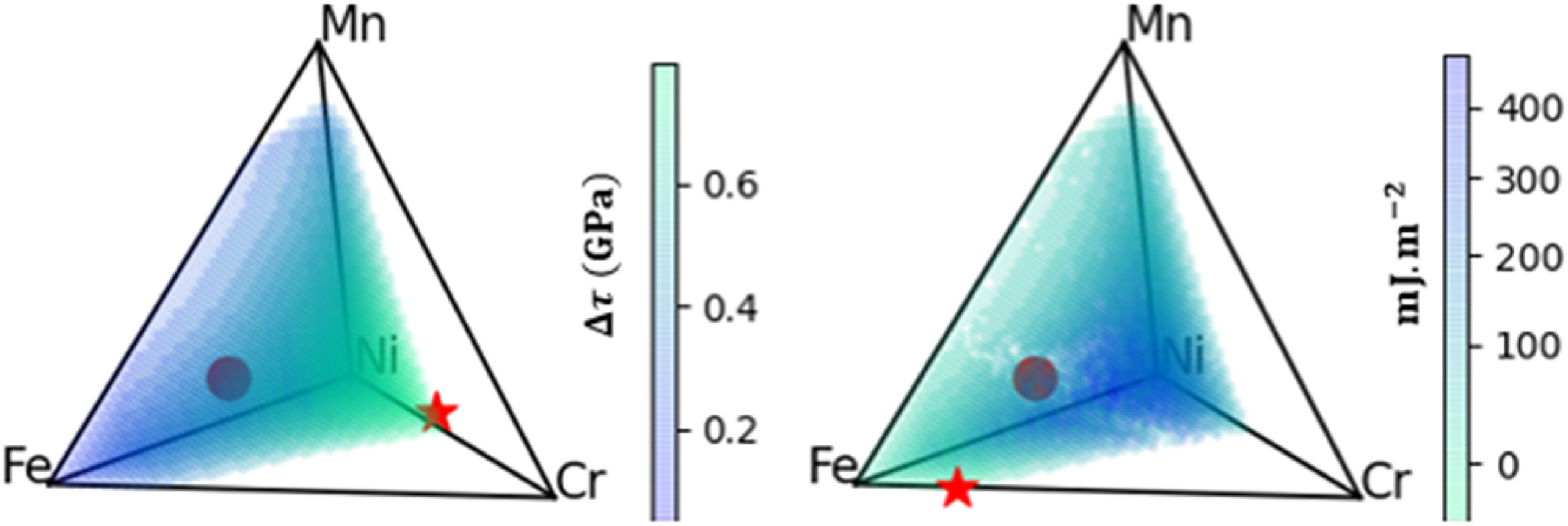
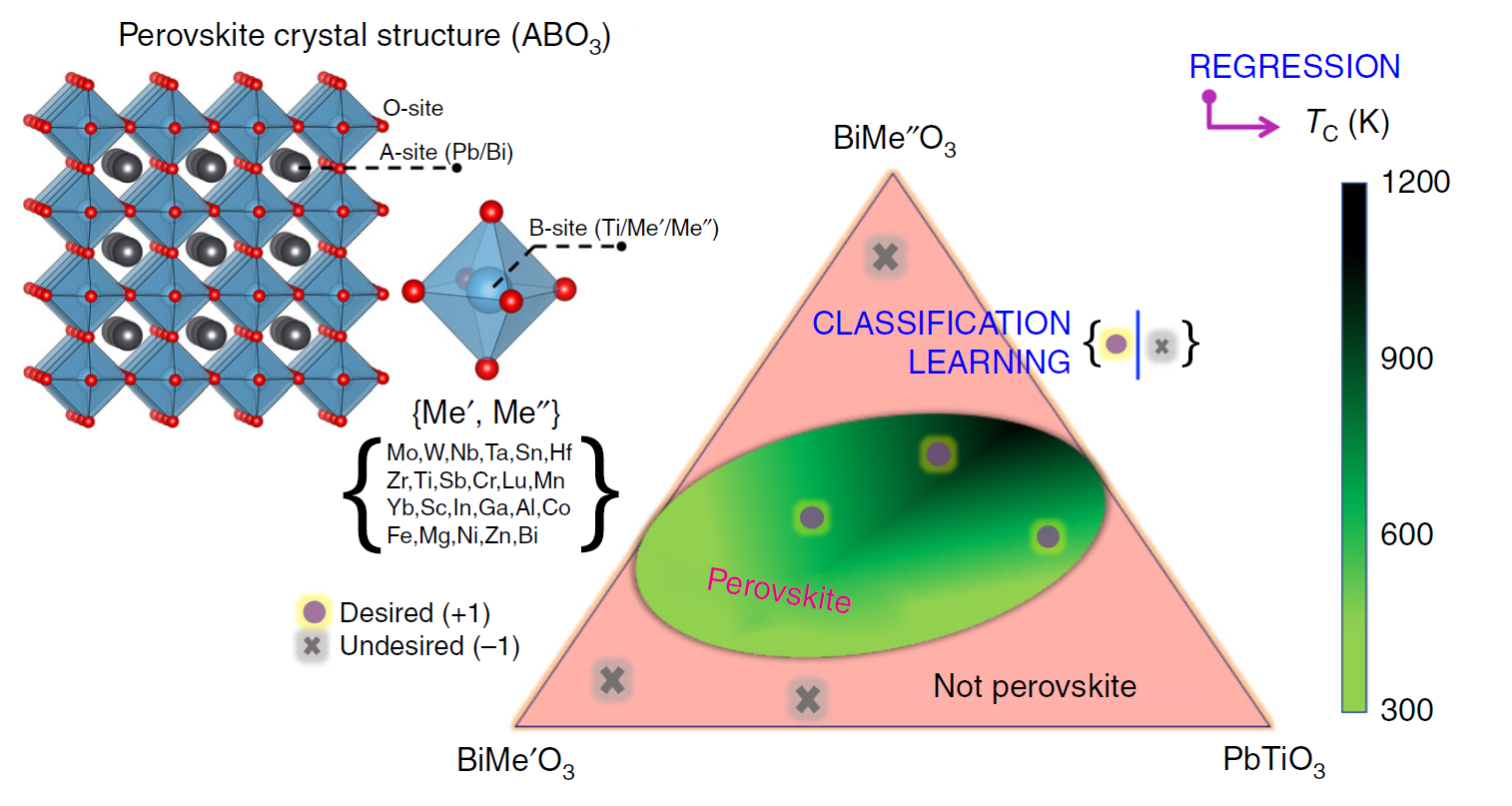
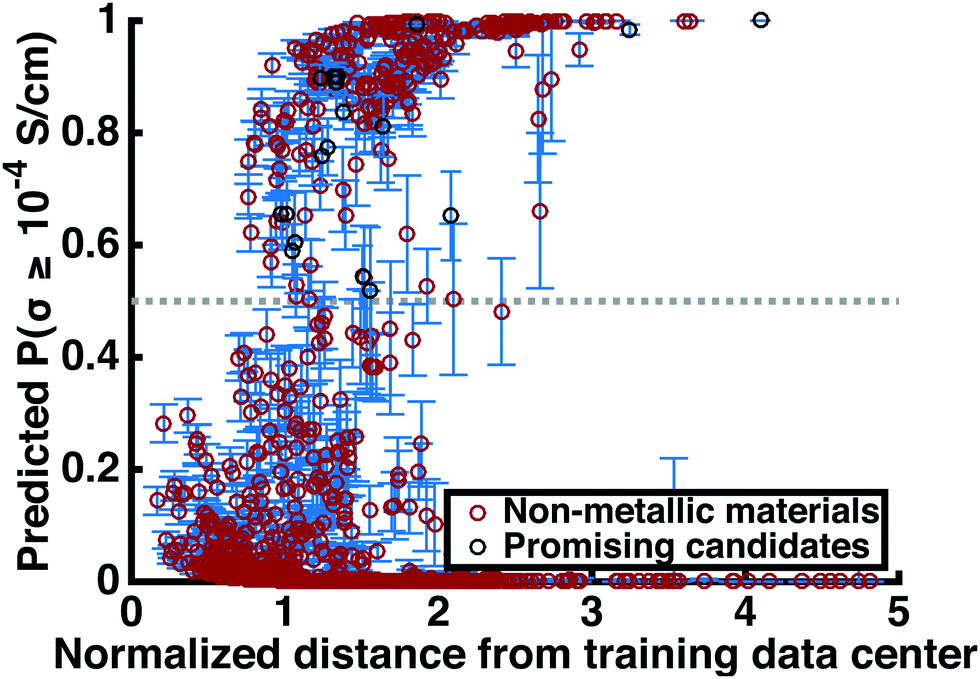
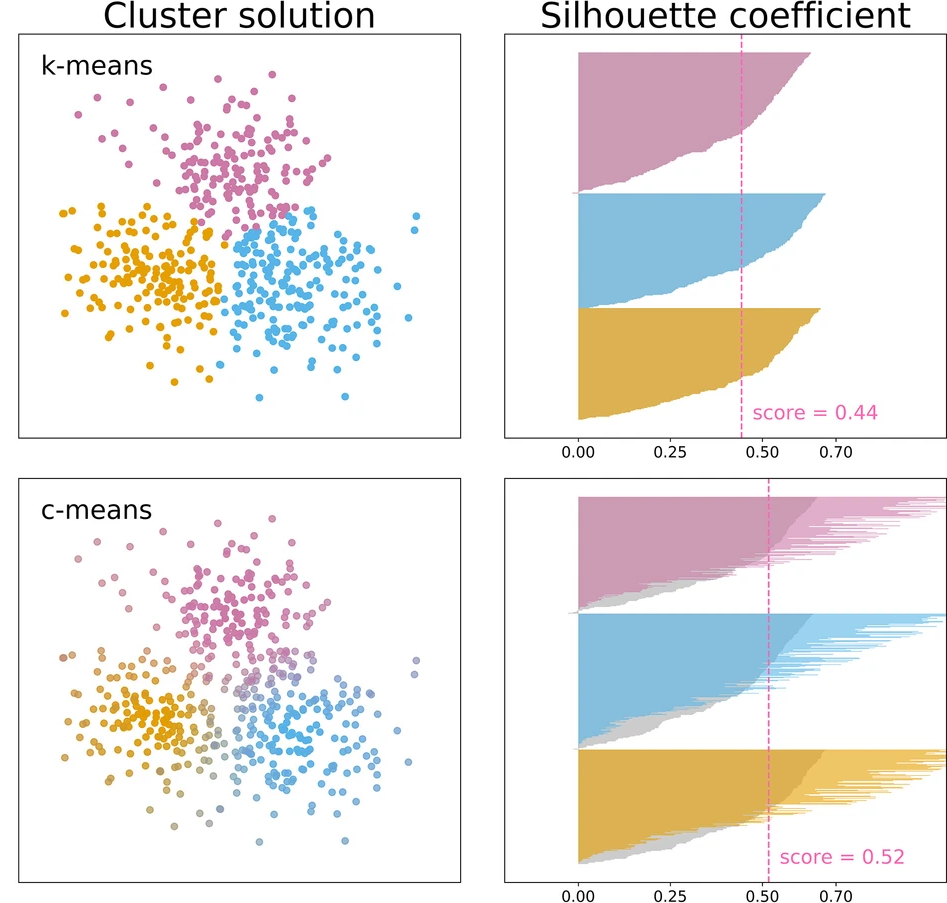


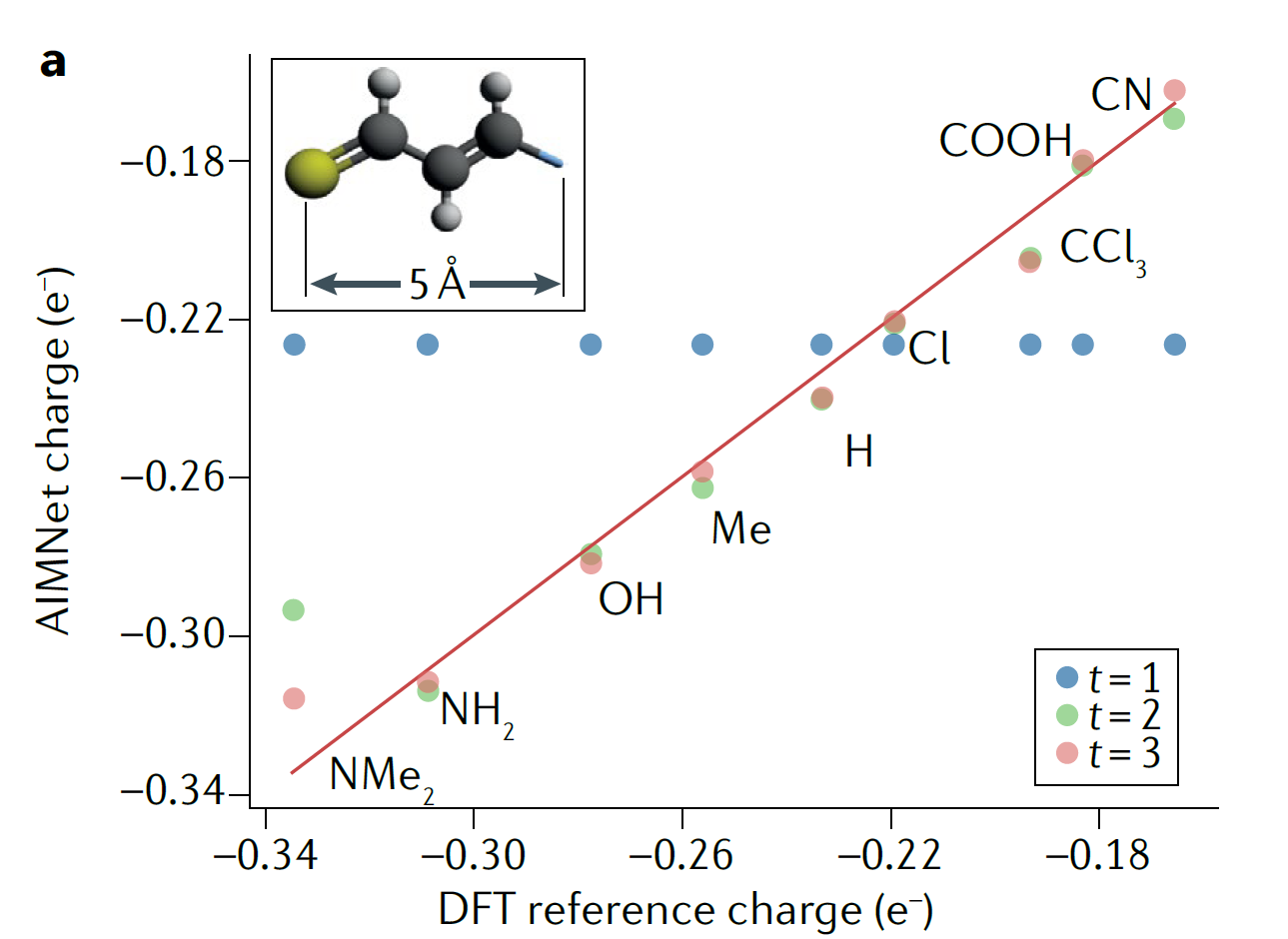
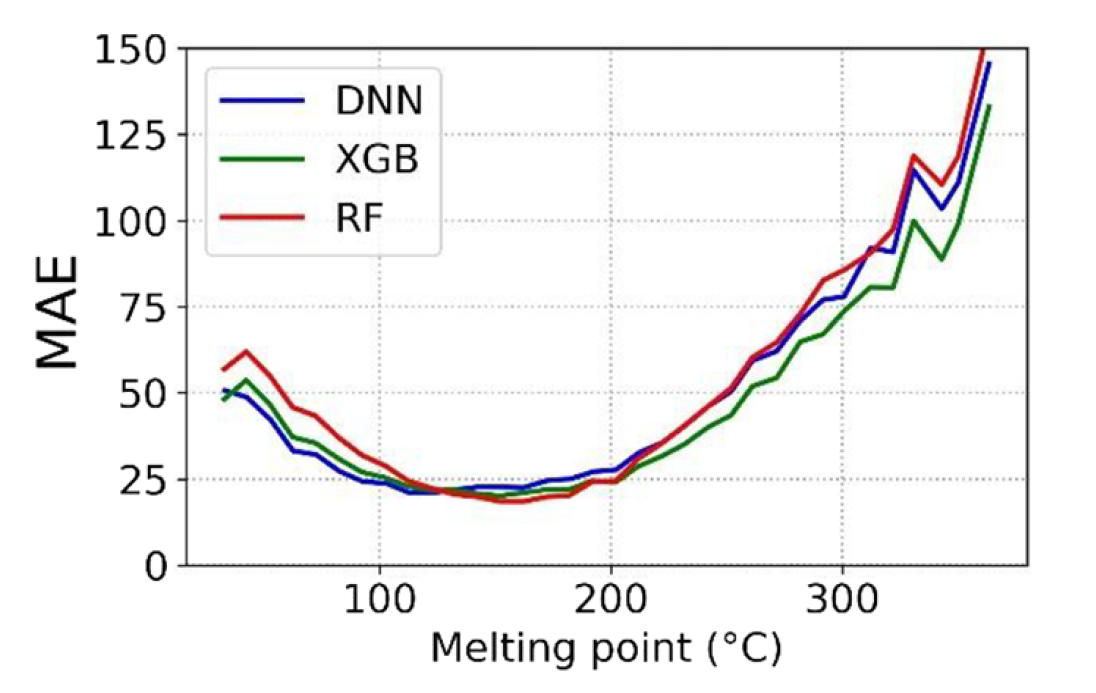
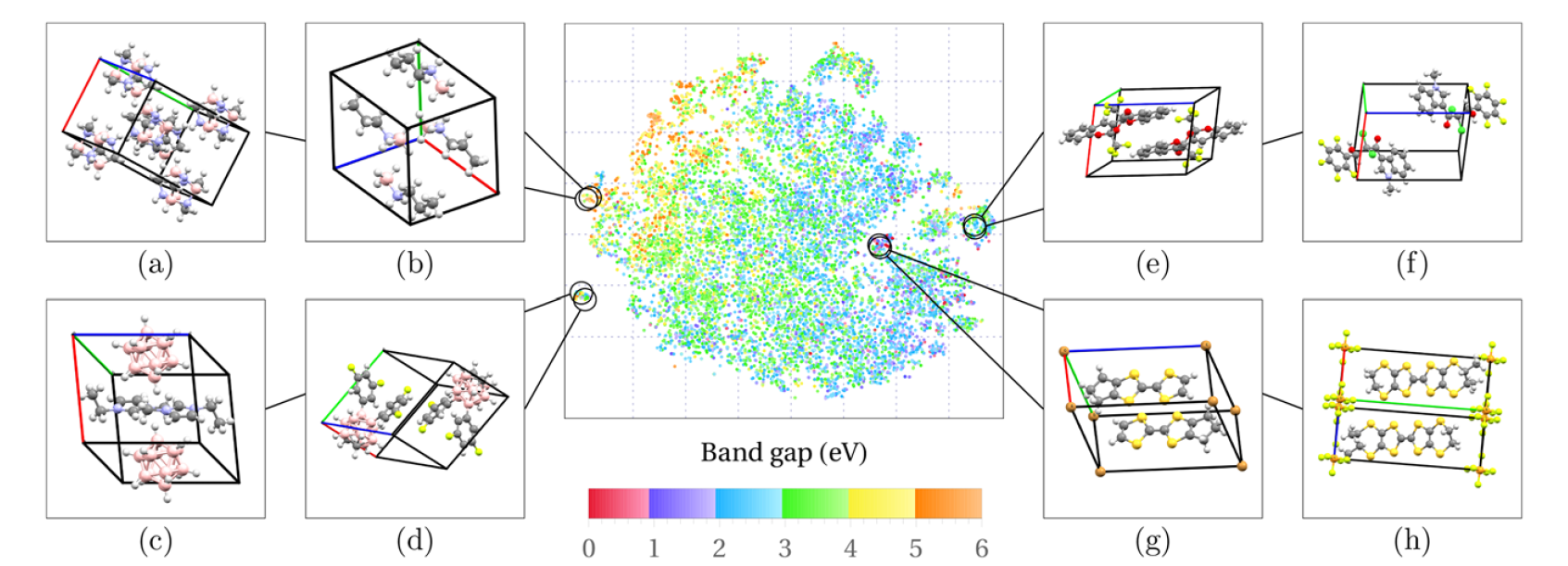
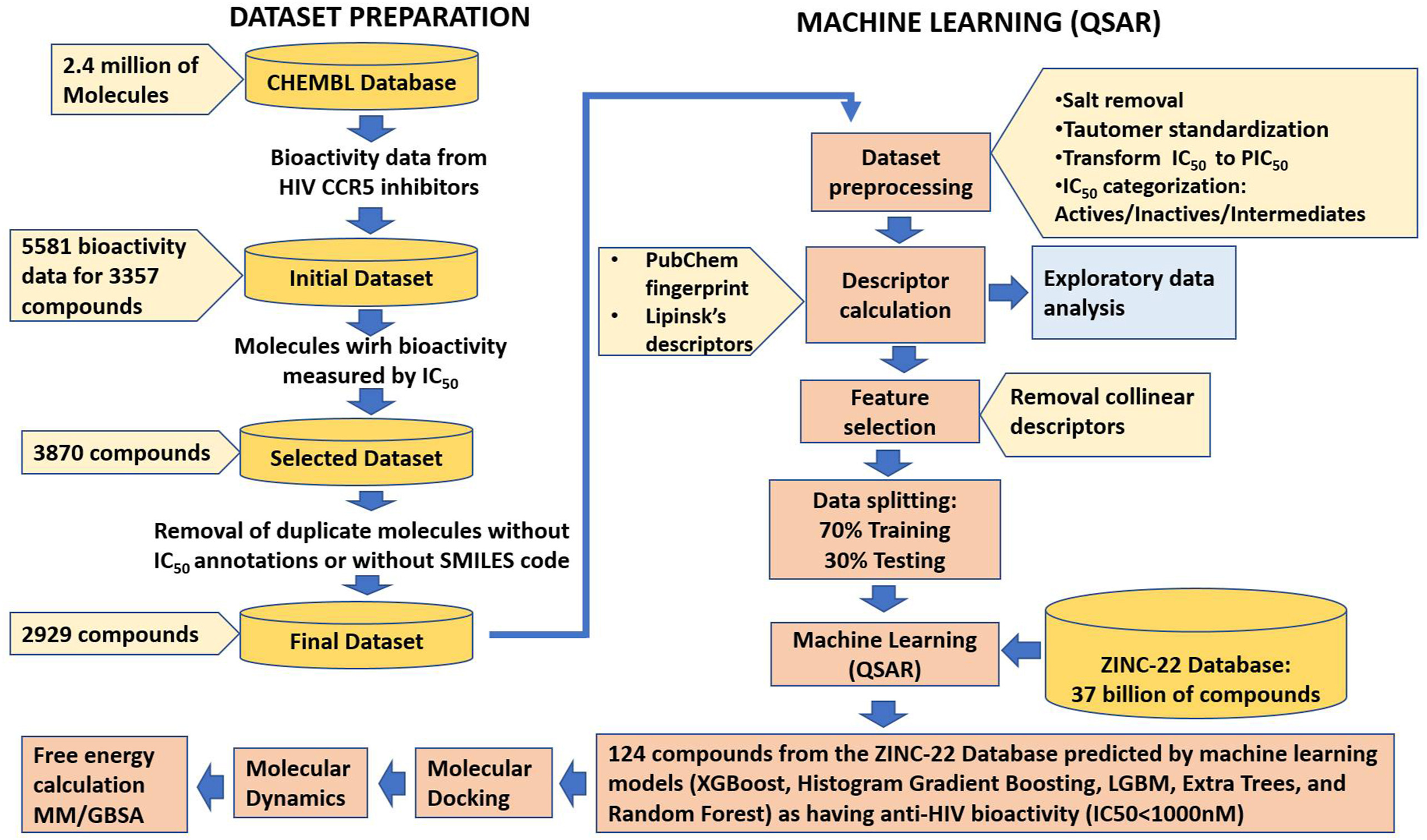
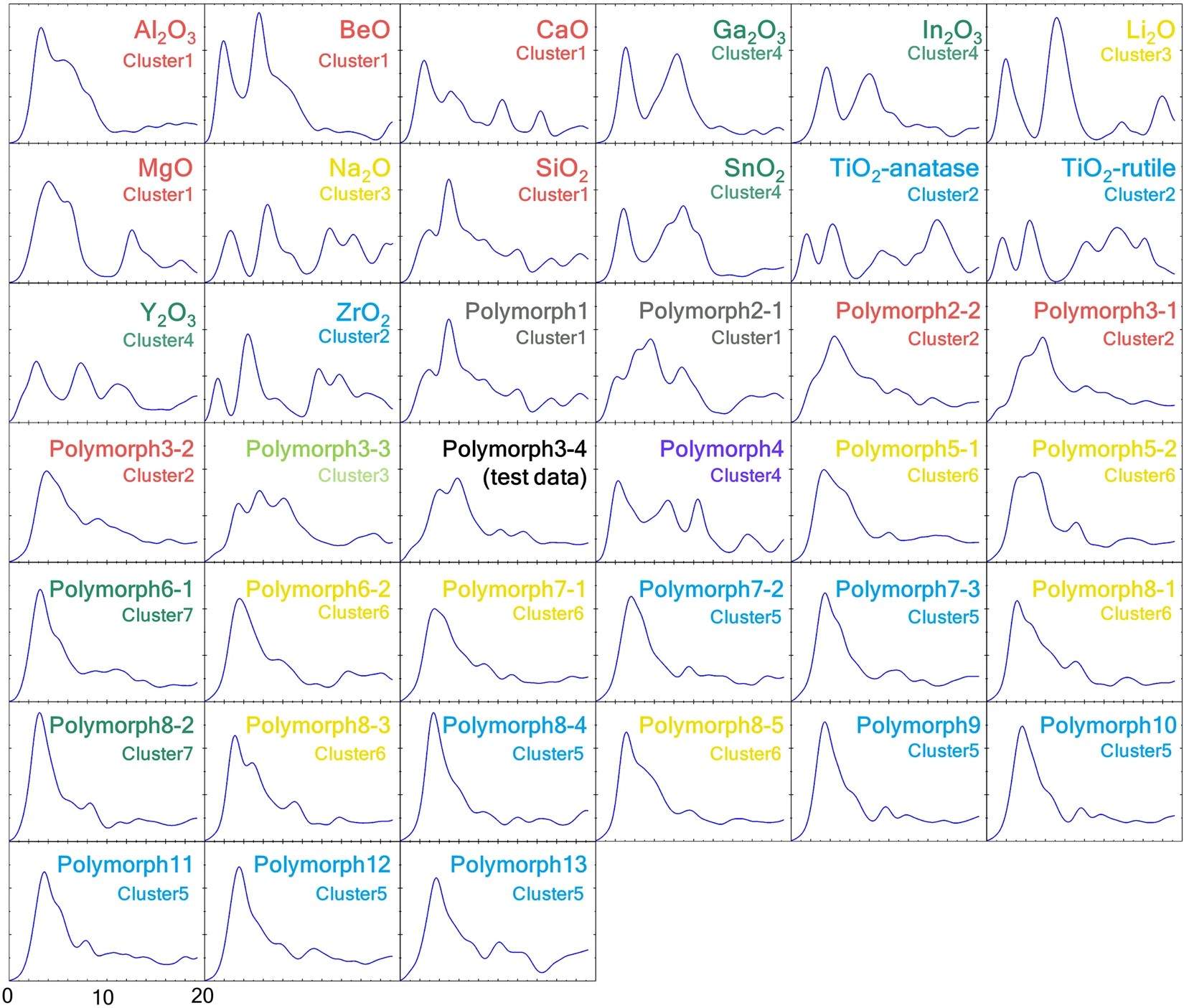
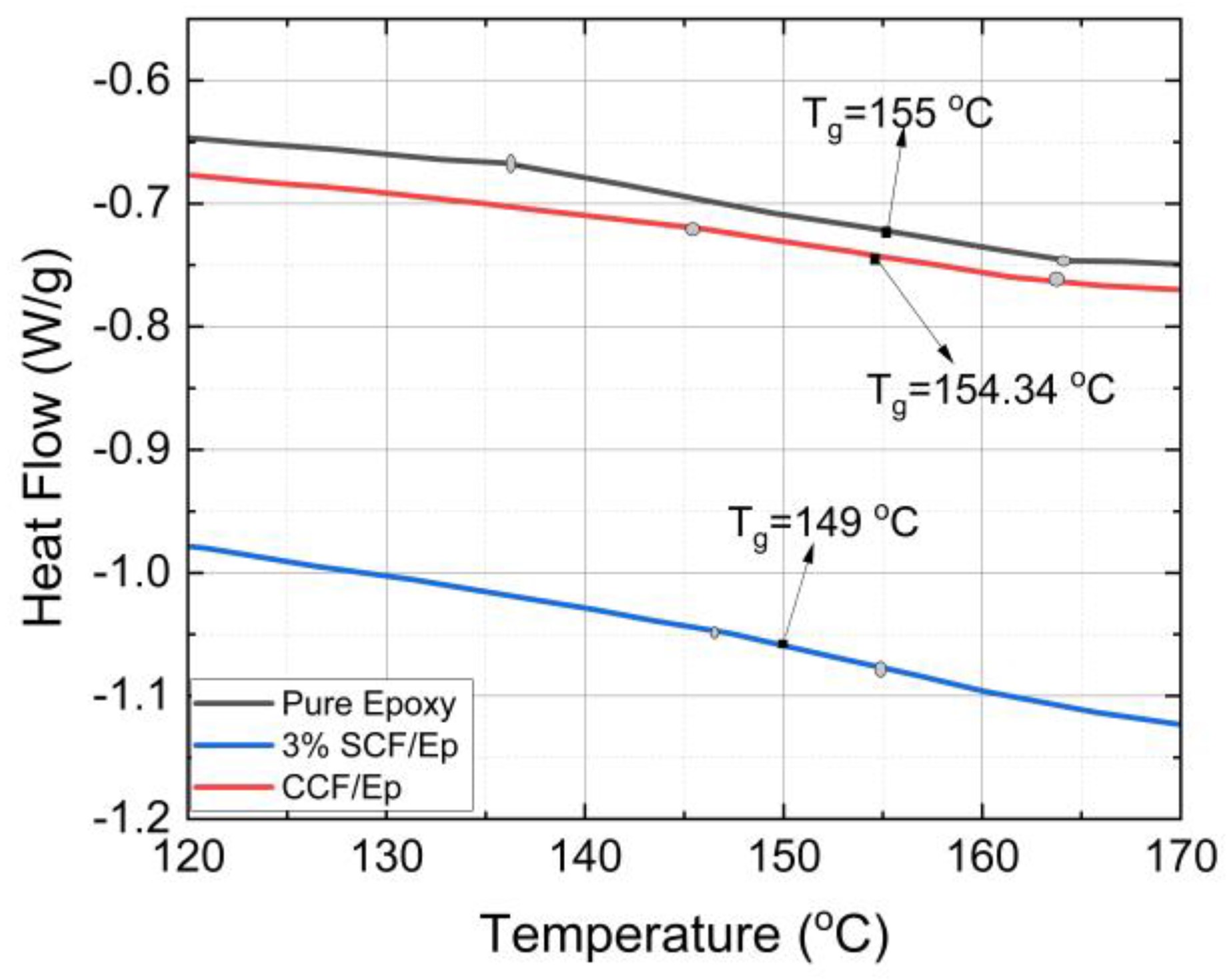
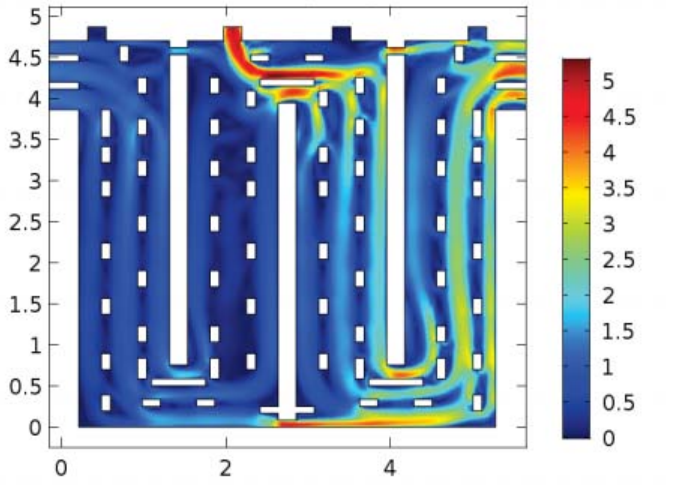
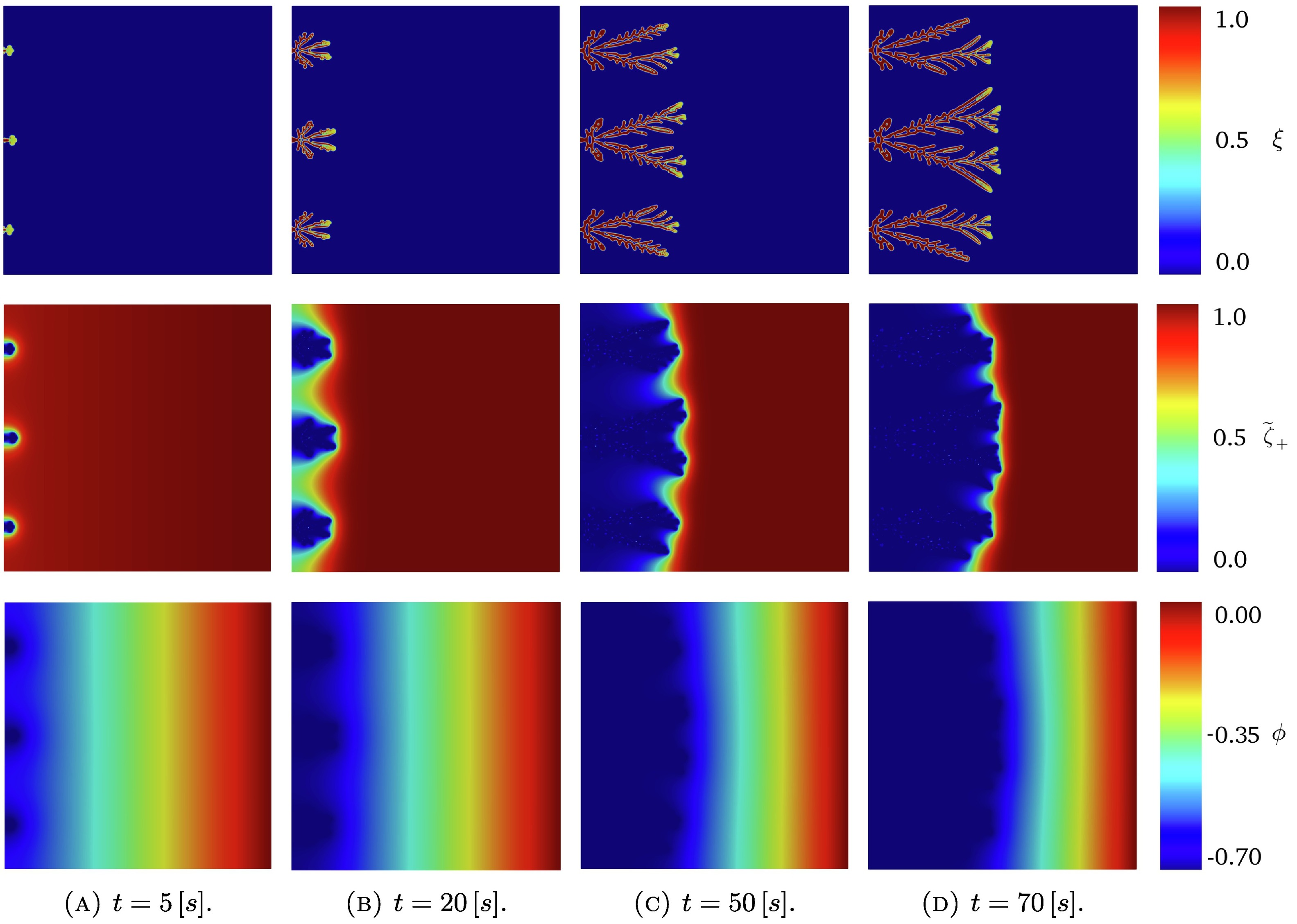
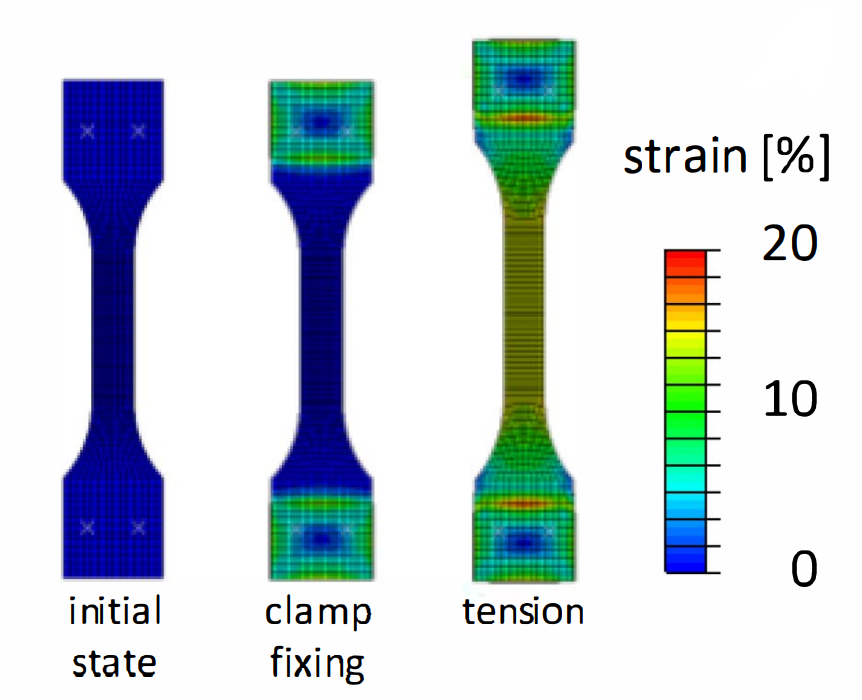
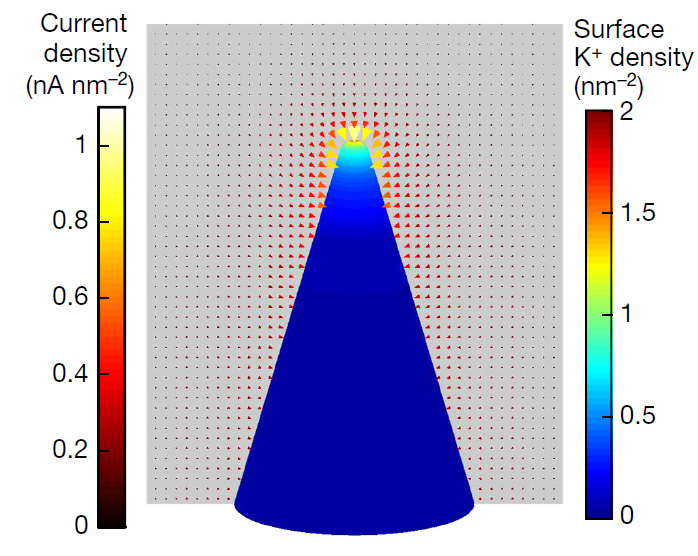
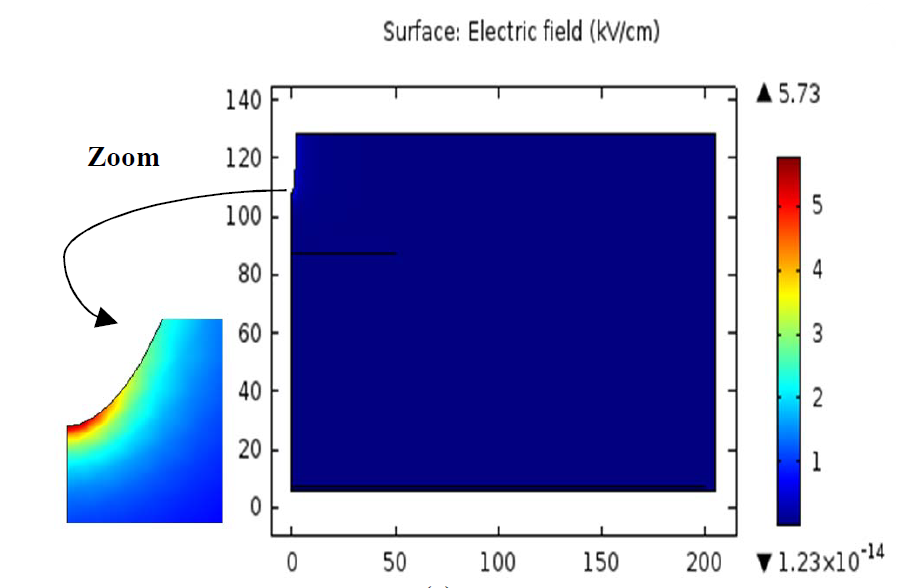
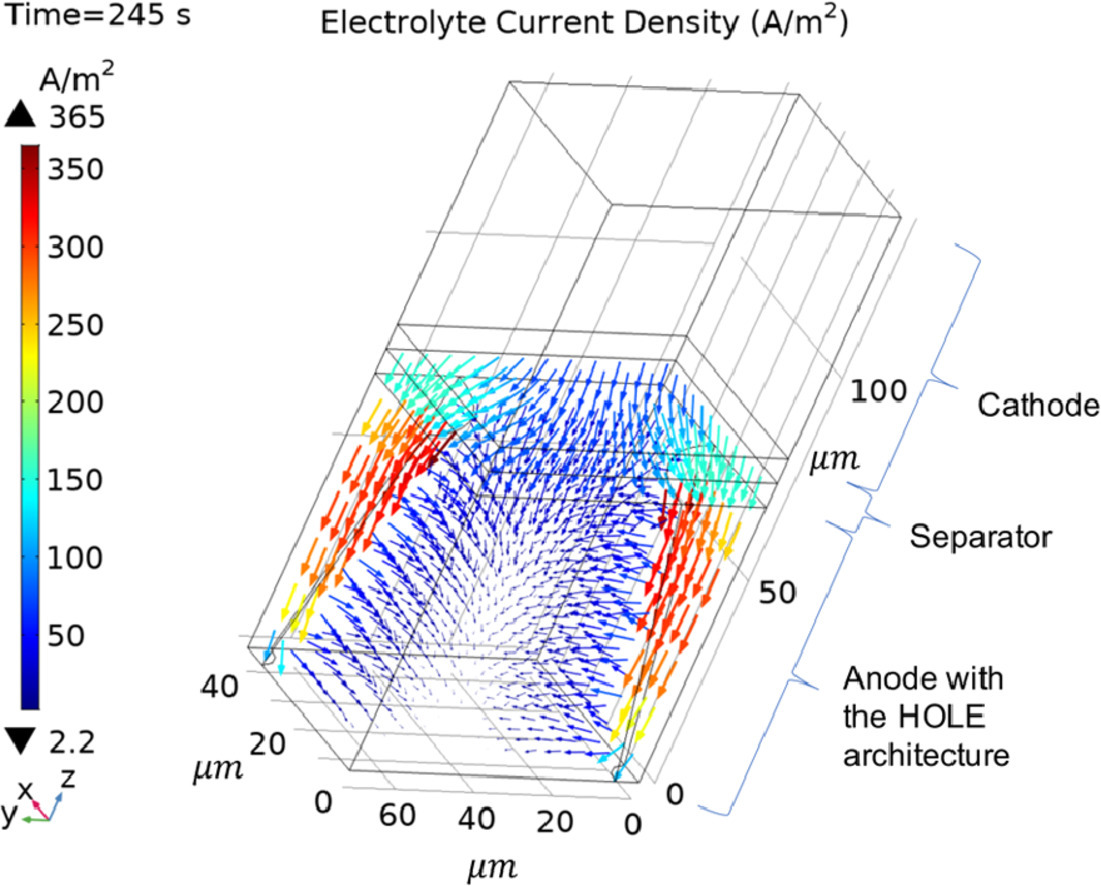
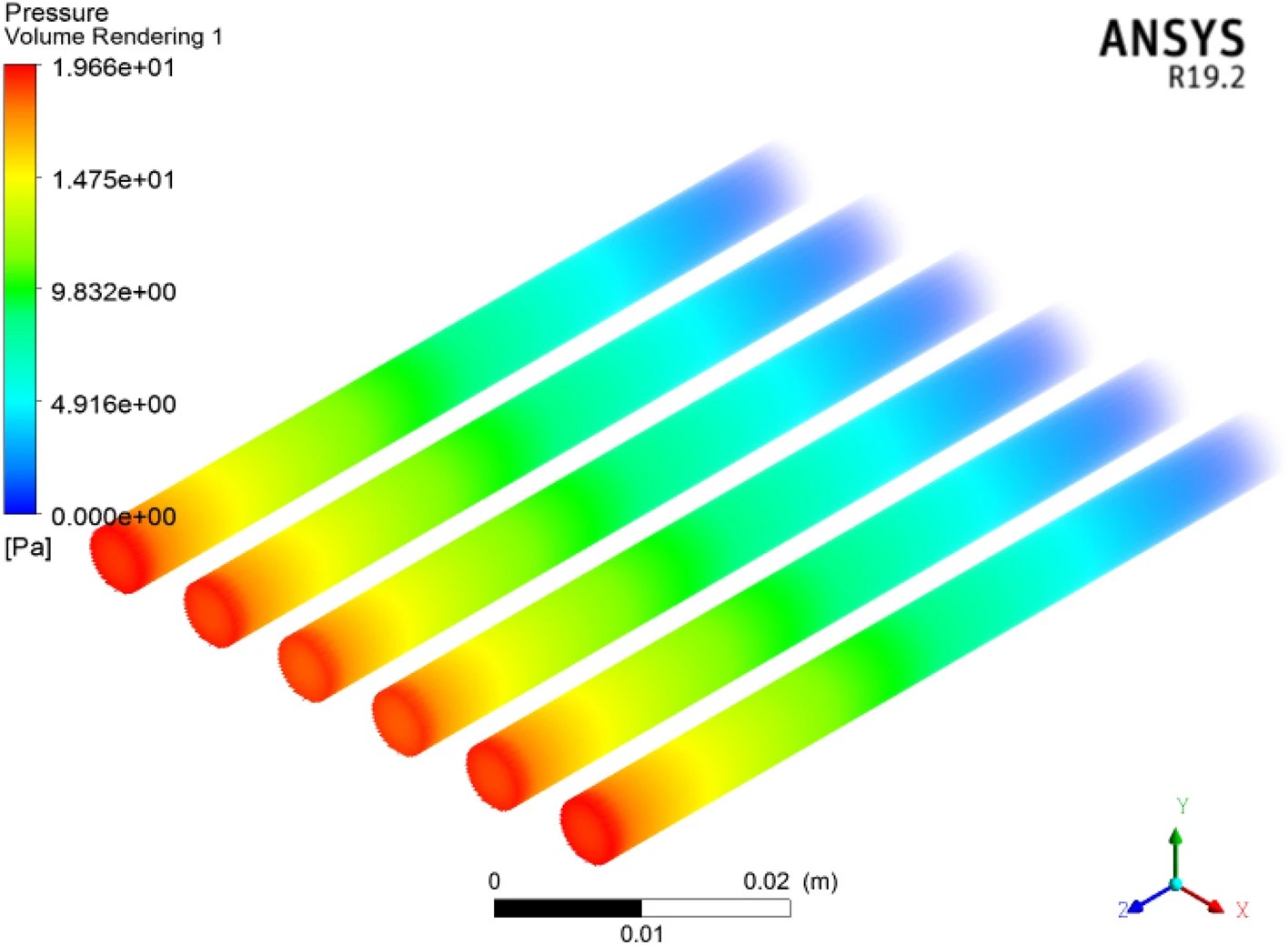
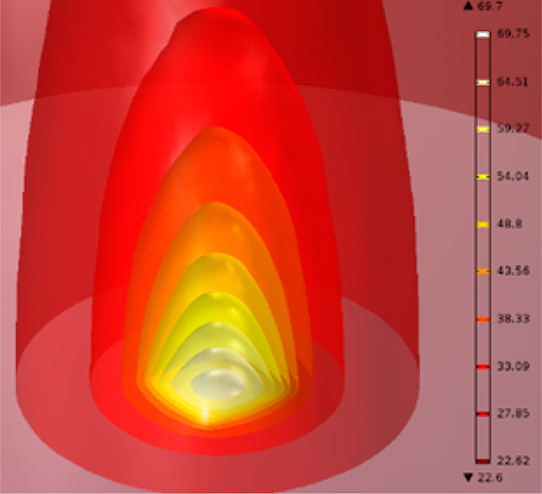
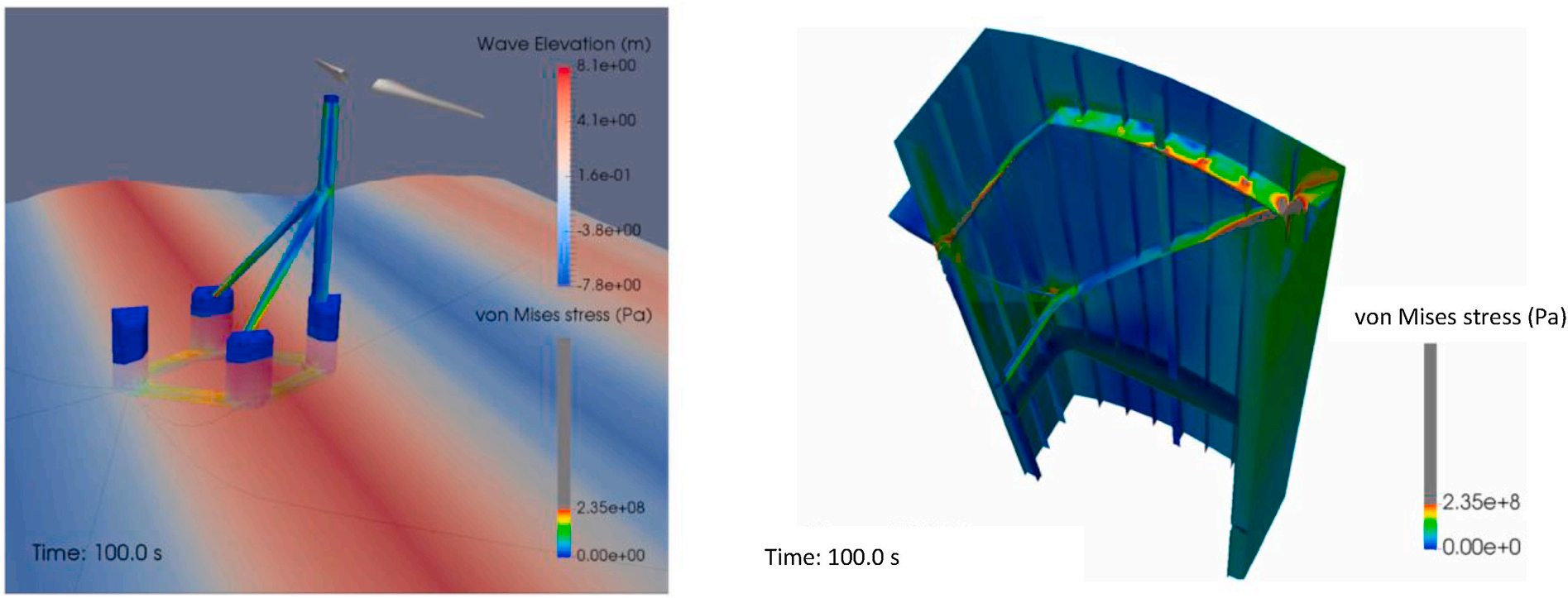
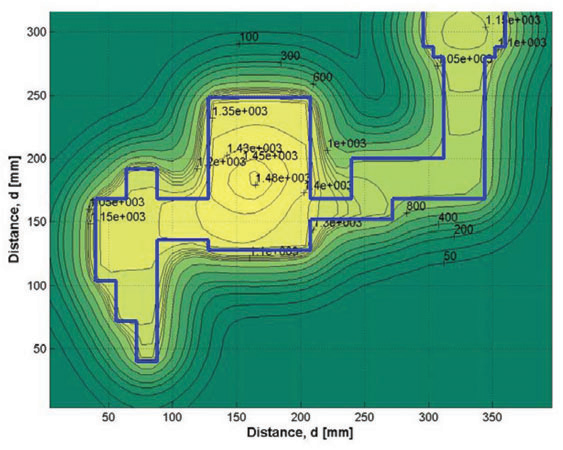
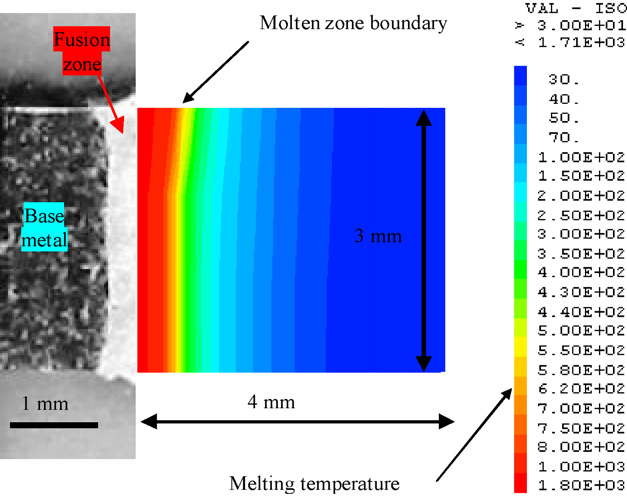
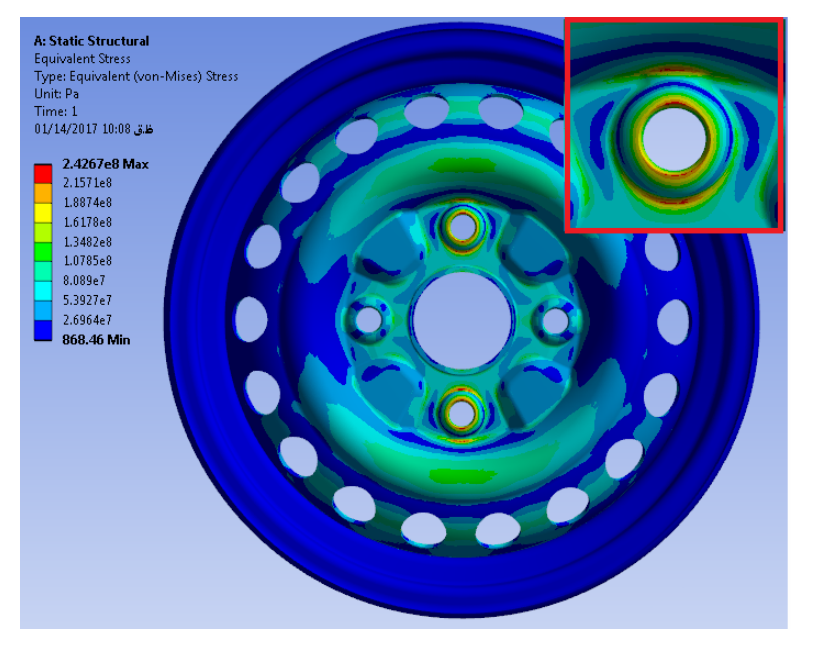
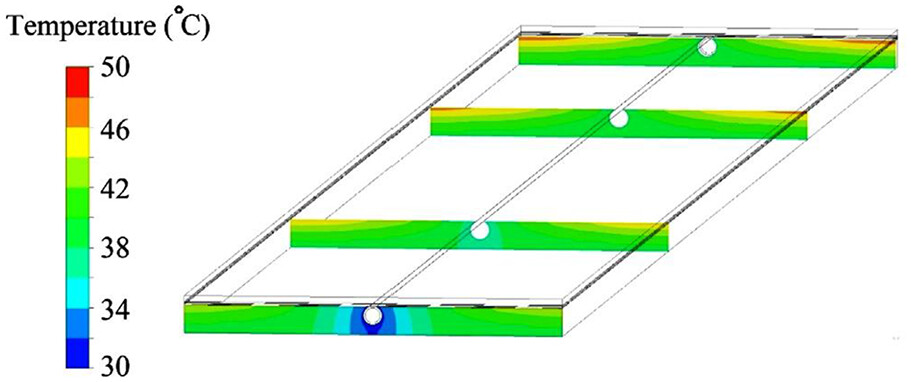
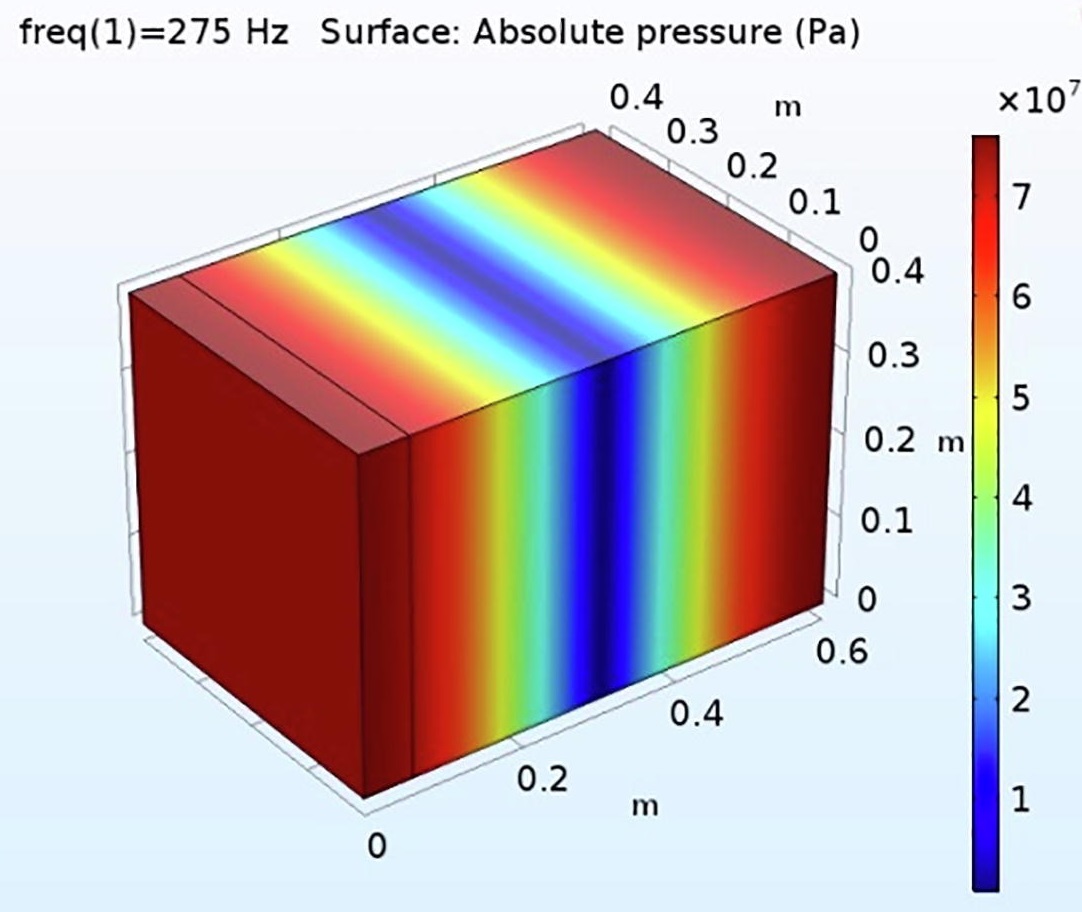
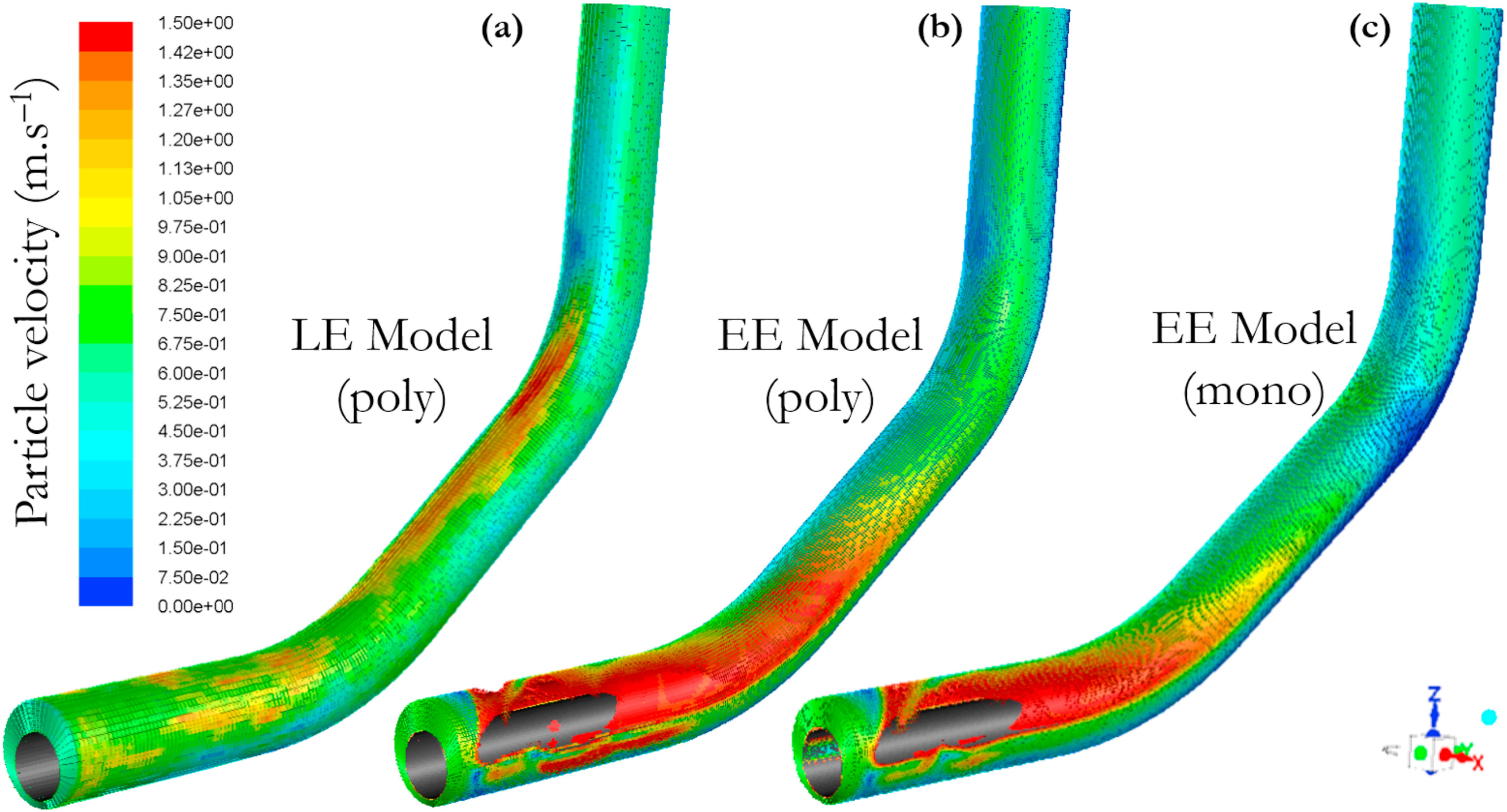
三维模型的搭建、网格划分、多相流体、传热模拟、传质模拟、电池温度分布、锂枝晶生长、电池容量衰减、阻抗分解、结构仿真(损伤、弯曲、碰撞、振动、断裂、静力分析、疲劳分析)、电化学仿真、力学仿真、电场仿真、光学仿真、热学仿真、磁场分布、多场耦合、电磁耦合、流固耦合、热固耦合、电磁波吸收



 提交需求
多种计算解决方案,量需定制
提交需求
多种计算解决方案,量需定制
 细节沟通
专业硕博顾问团队1V1沟通
细节沟通
专业硕博顾问团队1V1沟通
 付款确认
多种优惠政策,优质价廉
付款确认
多种优惠政策,优质价廉
 数据交付
全程反馈进度,先确认后交付
数据交付
全程反馈进度,先确认后交付
 结果验收
标准化流程,售后无忧
结果验收
标准化流程,售后无忧