本次介绍一篇于8月12日在《美国化学会志》上发表的突破性研究。该研究提出了一种创新的氨合成方法,通过融合Mo2CTx MXene与多种非贵金属元素(钴Co、镍Ni和铼Re),采用双活性位点策略和氢溢流效应,降低能耗并减少CO2排放,实现在更温和条件下的高效氨合成。本次介绍内容分为两部分:
氨是化学工业的基础,也是清洁能源的潜在载体。传统的Haber-Bosch(HB)法合成氨既耗能又导致大量CO2排放。为了降低对化石燃料的依赖并减少碳排放,开发在温和条件下有效的催化剂对电解驱动的HB法合成氨至关重要。福州大学江莉龙教授及合作者通过结合Mo2CTx MXene和多种非贵金属元素(Co、Ni和Re),利用双活性位点策略和氢溢流效应,以期在较低的温度和压力下实现高效氨合成,该篇文章以“Precious-Metal-Free Mo-MXene Catalyst Enabling Facile Ammonia Synthesis Via Dual Sites Bridged by H-Spillover”为题,发表在《Journal of the American Chemical Society》顶刊上。
https://doi.org/10.1021/jacs.4c03998
活性问题:非贵金属(NPM)催化剂在氨合成中的活性通常较低,因为它们在N2解离和NH3脱附的能量障碍之间存在反向关系。
能量障碍:在传统的过渡金属(TM)催化剂上,N2解离的能量障碍与NHx(x = 1−3)的脱附能量之间存在火山曲线关系,这限制了催化剂的性能。
条件匹配问题:电解水产生的氢气压力与氨合成催化剂所需的压力不匹配,这限制了电解驱动的Haber-Bosch(eHB)过程的效率和成本效益。
双活性位点策略:通过将非贵金属(如Co、Ni和Re)与Mo2CTx MXene结合,利用双活性位点策略,其中一个位点负责N2的激活,另一个位点负责H2的激活。
氢溢流效应:利用氢溢流,即活化的氢原子从非贵金属转移到Mo2CTx表面,促进N2的氢化和NH2的生成。
表面功能化和电子转移:通过部分去除Mo2CTx表面的氧官能团(Tx),暴露出Mo4+位点,这些位点能够强有力地激活N2。同时,非贵金属位点上的电子转移有助于促进H2的激活和NH2的脱附。
理论计算与实验相结合:使用密度泛函理论(DFT)计算来模拟氨合成的反应路径,结合实验观察到的现象,优化催化剂的设计。
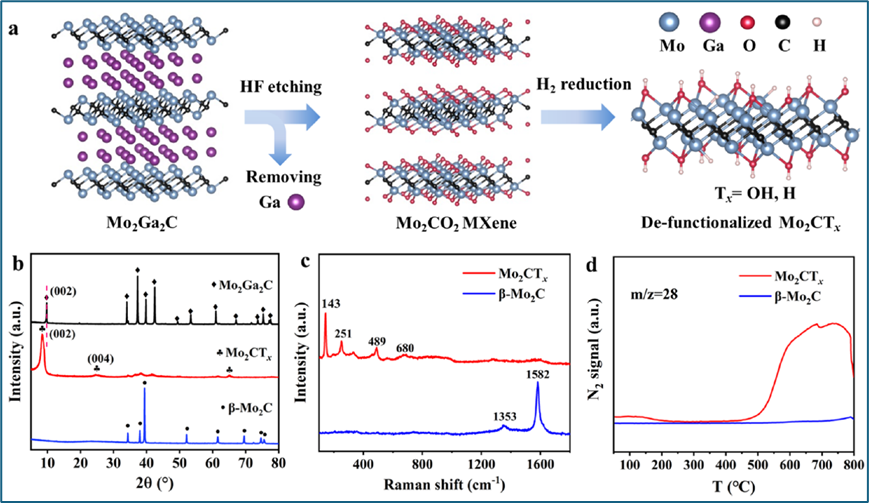
Mo2CTx MXene是通过氢氟酸刻蚀Mo2Ga2C前驱体并用碱液洗涤来合成的,具有均匀分散的Mo、C和O元素。其结构通过XRD和SEM分析显示为手风琴状多层二维形态,拉曼光谱表明其表面主要由氧物种终止。H2-TPR和N2-TPD-MS测试结果揭示了Mo2CTx在还原条件下氧官能团可部分转化为H或OH基团,同时表现出对N2的良好吸附性。这些特性使移除官能团的Mo2CTx成为一种有潜力的氨合成催化剂。
图1. Mo2CTx催化剂的合成与表征。(a) 2D Mo2CTx MXene合成过程示意图,(b) Mo2CTx、Mo2Ga2C和β-Mo2C的XRD图谱,(c) Mo2CTx和Mo2C的拉曼光谱,(d) 使用在线质谱仪记录的Mo2CTx和β-Mo2C的N2-TPD曲线。
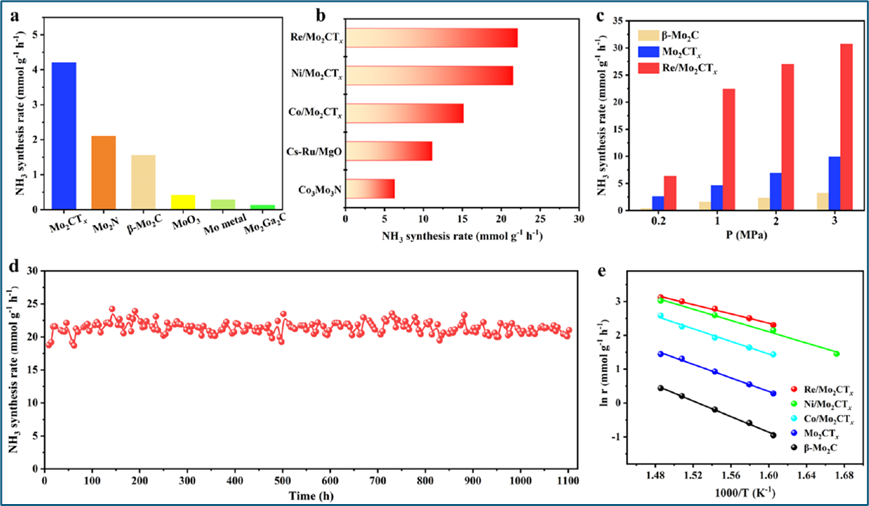
通过在25% N2和75% H2的气氛下,以60000 mL/g·h的空速对新制备的催化剂进行氨合成性能评估。结果显示,移除官能团的Mo2CTx在400°C和1 MPa的条件下,氨合成速率是β-Mo2C的2.8倍。通过在Mo2CTx上负载NPMs,如Co、Ni和Re,进一步显著提高了氨合成速率,其中Re/Mo2CTx的速率在非贵金属催化剂中是最高的,甚至超过了典型的贵金属Ru基催化剂。动力学分析表明,Mo2CTx和NPMs/Mo2CTx催化剂的表观活化能较低,表明N2在这些催化剂上的活化较为容易。此外,Mo2CTx和NPMs/Mo2CTx的N2吸附和活化能力较高,氨合成反应的速率限制步骤可能从N2的直接解离转移到N−H键的形成。
图2.氨合成的催化性能。(a) 不同基于钼的催化剂和(b) 400 ℃、1 MPa下NPM介导的Mo2CTx样品以及参照催化剂的氨合成速率,(c) 400 ℃下不同催化剂的氨合成速率与反应压力的关系,(d) 400 ℃、1 MPa下Re/Mo2CTx的长期稳定性测试,(e) NPMs/Mo2CTx、Mo2CTx和β-Mo2C的阿累尼乌斯图。
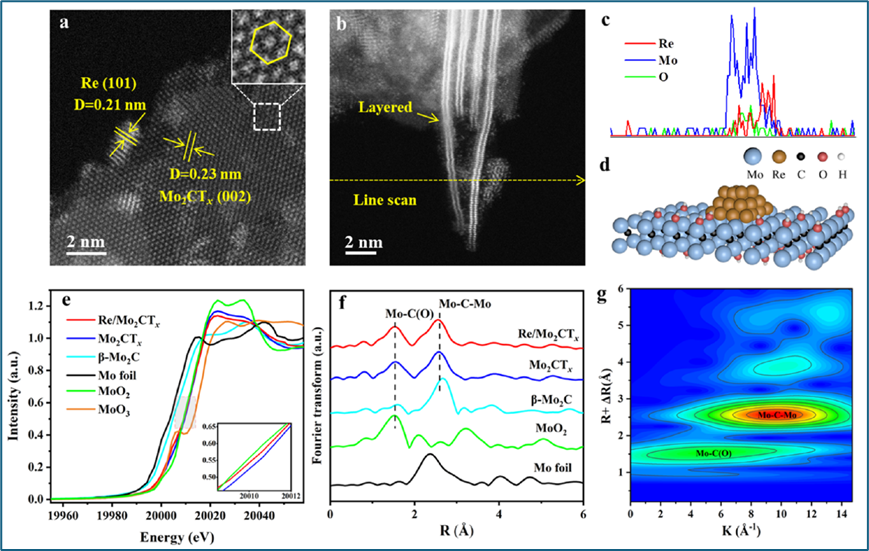
采用HRTEM等表征手段来确定Re/Mo2CTx催化剂的结构特性。HRTEM图像显示,Re颗粒在Mo2CTx中高度分散,平均粒径约为2.3nm。经过长期稳定性测试后,Re颗粒的平均尺寸仅从2.3 nm增加到2.5 nm,表明Re/Mo2CTx在氨合成中具有高稳定性。利用AC-HAADF-STEM清晰地展示了Re金属和Mo2CTx的晶格间距。EXAFS光谱进一步揭示了Mo2CTx的配位结构,并且通过小波变换分析确认了Re/Mo2CTx中Mo−C(O)和Mo−C−Mo的配位。
图3. Re/Mo2CTx的几何和电子结构。(a, b) Re/Mo2CTx的代表性AC-HAADF-STEM图像,(c)线扫描结果,(d) 提出的Re/Mo2CTx模型,(e) Re/Mo2CTx和参照样品的Mo K边XANES归一化以及 (f) k2加权傅里叶变换EXAFS光谱在r空间的表现,(g) Re/Mo2CTx催化剂k2加权EXAFS信号的小波变换。
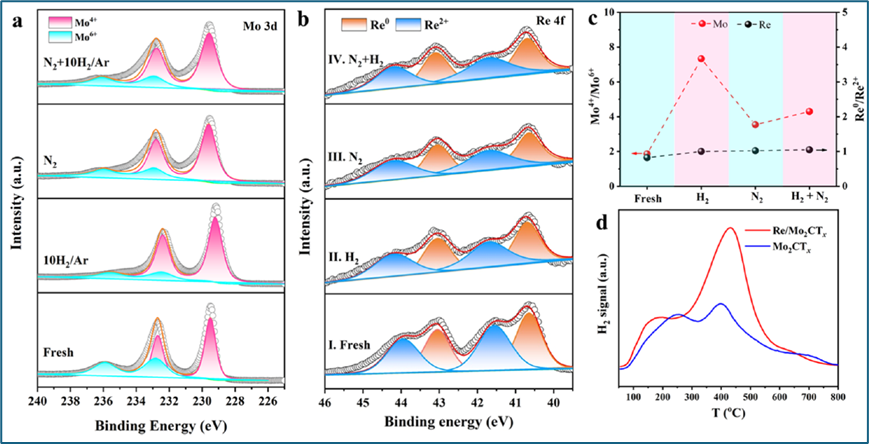
通过准原位XPS实验研究了Re和Mo2CTx在氨合成中对N2和H2激活的作用。实验中,Re/Mo2CTx在不同气氛下处理,并通过XPS光谱观察了Mo 3d和Re 4f的演变。原始的Re/Mo2CTx中Mo4+和Mo6+的比例分别为65%和35%,在经过H2预处理后,Mo4+的比例增加,表明H2还原去除表面氧物种,暴露出更多的Mo4+位点。随后在N2气氛中的处理导致Mo4+/Mo6+比例下降,说明Mo4+位点可以激活N2,形成Mo6+-N物种。在N2-H2气氛中的处理又使Mo4+/Mo6+比例上升,表明部分Mo6+-N物种通过氢化反应转化为NH2并恢复Mo4+位点。实验还发现,Re物种在不同气氛下的数目没有显著变化,表明Re物种在N2激活中不是主要参与者。此外,通过在线质谱监测H2和N2信号,发现Mo2CTx上的H2解吸量在通入H2和N2时减少,表明H2和N2之间存在竞争吸附。DFT计算和同位素交换实验进一步证实了Mo2CTx上N2的优先吸附和Re/Mo2CTx上的H溢流现象。
图4. Re/Mo2CTx的N2和H2激活的双活性位点。在400 °C下2小时不同气体环境下Re/Mo2CTx的准原位XPS光谱(a) Mo 3d和(b) Re 4f,(I) 原始样品,(II) 10%H2/Ar,(III) 2.5%N2/Ar,以及(IV) 2.5%N2−7.5%H2/Ar,(c) 分别从(a)和(b)得出的Mo4+/Mo6+和Re0/Re2+的比率,(d) 在Mo2CTx和Re/Mo2CTx上N2和H2气体共吸附后TPD实验中H2解吸的信号。
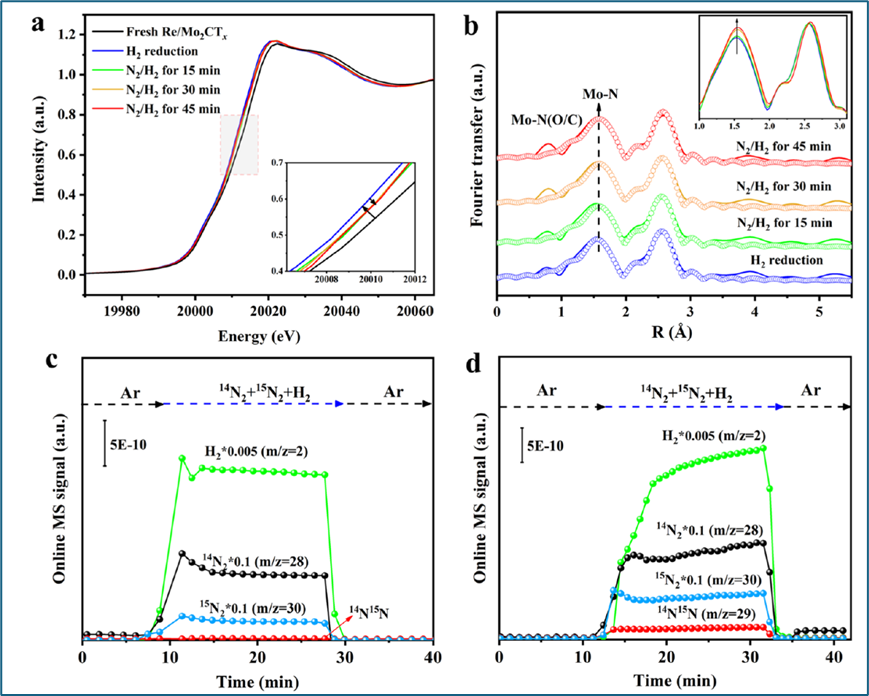
使用自制的反应池和先进的原位技术,对Re/Mo2CTx催化剂上氨合成的反应过程进行了研究。Mo K边XANES光谱显示,在H2还原后,Mo的价态降低,而在N2-H2气氛中反应进行时,Mo的价态增加,这归因于Mo位点向N2的反键π*轨道提供电子。原位Mo K边EXAFS光谱表明,随着反应进行,Mo−C(N)的配位数增加,而Mo−C−Mo的配位数保持不变,这表明Mo位点在N2激活和形成Mo−N键中起着关键作用。通过同位素交换实验,发现Mo2CTx和Re/Mo2CTx催化剂上的N2解离容易,且反应速率限制步骤从N2解离转移到了N−Hx的形成。此外,Re/Mo2CTx上的H2解吸量高于Mo2CTx,表明Re的加入提供了额外的H2激活位点,并可能发生了H溢流现象。实验还发现,Re/Mo2CTx上NH3的生成和脱附更为容易,这与Re在促进H2激活和H溢流中的作用有关,从而增强了NH3合成的性能。
图5. Re/Mo2CTx上的构效关系。(a) 原位归一化的Mo K边XANES和(b) 400 °C下10%H2/Ar中45min和不同时间在线的2.5%N2−7.5%H2/Ar中Re/Mo2CTx催化剂的k2加权傅里叶变换Mo K边EXAFS光谱及其相应的曲线拟合结果。(c) β-Mo2C和(d) Re/Mo2CTx在400 °C下引入14N2-15N2−H2后的质谱信号,体积比为14N2/15N2/H2= 2:1:6。
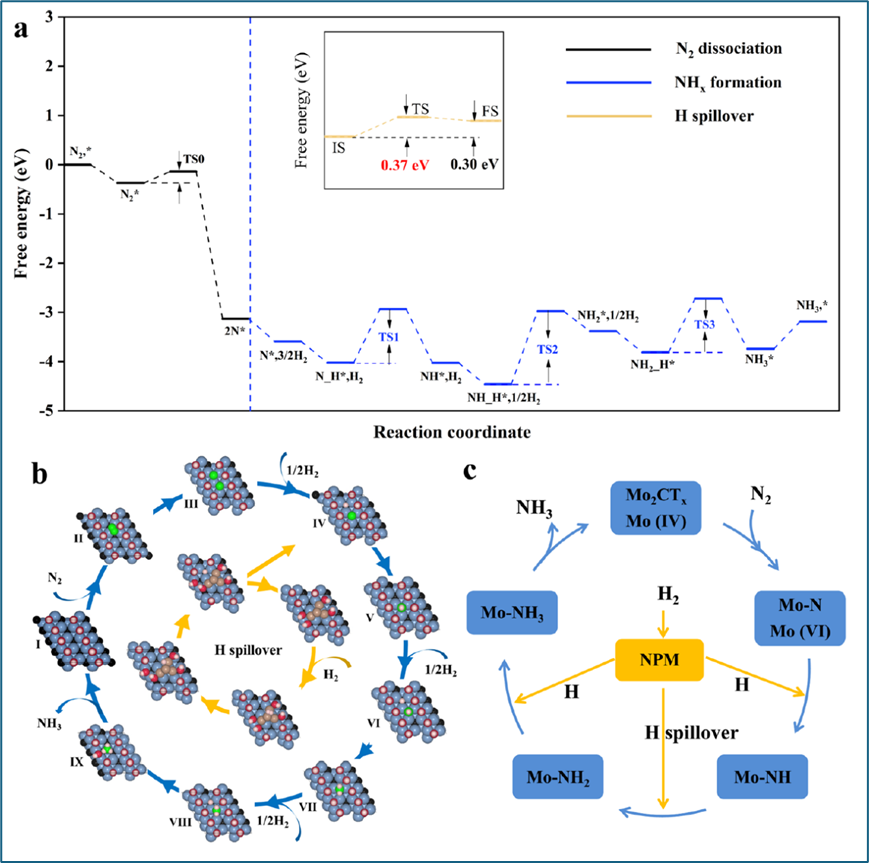
通过DFT计算,研究者探究了Re/Mo2CTx催化剂上氨合成反应的机理。构建的Mo2COH模型显示,N2在Mo2COH表面吸附稳定,且Mo位点能有效活化N2,避免N2中毒。实验数据和计算结果共同证实了Mo位点在N2活化中的关键作用,并且N2活化可能不是氨合成的速率限制步骤。Re原子团的引入显著提升了催化性能,H2分子在Re上解离后,通过低能量势垒的H-溢流,为Mo2CTx表面提供了丰富的H源,促进了N−H键和NH2的形成。这些发现表明,Re/Mo2CTx催化剂通过协同效应,实现了在温和条件下的高效氨合成。
图6.反应机理的理论计算。(a) 计算Mo2CTx上NH3合成的自由能路径和Re到Mo2CTx的H溢流路径,(b) NH3合成的关键基元步骤(I−IX)的结构显示在外圈,Re到Mo2CTx的H溢流结构显示在内圈,(c) 双活性位点通过H溢流辅助NPM/Mo2CTx进行NH3合成的示意图。
目的:用于分析催化剂在不同反应条件下的表面电子结构变化。
过程:Re/Mo2CTx催化剂首先在400℃下用10%H2/Ar还原2小时,然后转移到分析室进行XPS测量。之后,样品在不同气体环境下进一步处理,每次处理后记录XPS光谱。
结果:通过分析Mo 3d和Re 4f的XPS光谱,研究者发现Mo4+和Mo6+的比例变化,这表明Mo2CTx表面的Mo4+位点在N2激活中起着关键作用。Re物种的数量变化不大,表明Re可能不直接参与N2的活化。
目的:原位XAFS技术用于实时监测催化剂在反应条件下的局部原子结构和价态变化,尤其是在NH3合成过程中。
过程:使用自制的反应池,在400℃下,对Re/Mo2CTx催化剂进行H2或N2-H2混合气体处理,并通过XANES和EXAFS实时监测其结构变化。
结果:原位Mo K-edge XANES光谱显示,在H2还原后Mo的价态降低,而在N2-H2气氛中反应进行时,Mo的价态增加。EXAFS光谱表明,随着反应的进行,Mo−C(N)的配位数增加,这与N2在Mo位点上的活化和随后的N−H键形成有关。
H溢流:一种现象,其中一个反应物(通常是氢)在催化剂的某个位点上被激活后,迁移到另一个位点参与反应。
准原位技术:接近原位条件的技术,但可能涉及某些处理步骤,如样品转移。
【高端测试,找华算】🏅 同步辐射 全球机时,三代光源,随寄随测!最快一周出结果,保证数据质量!


声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!