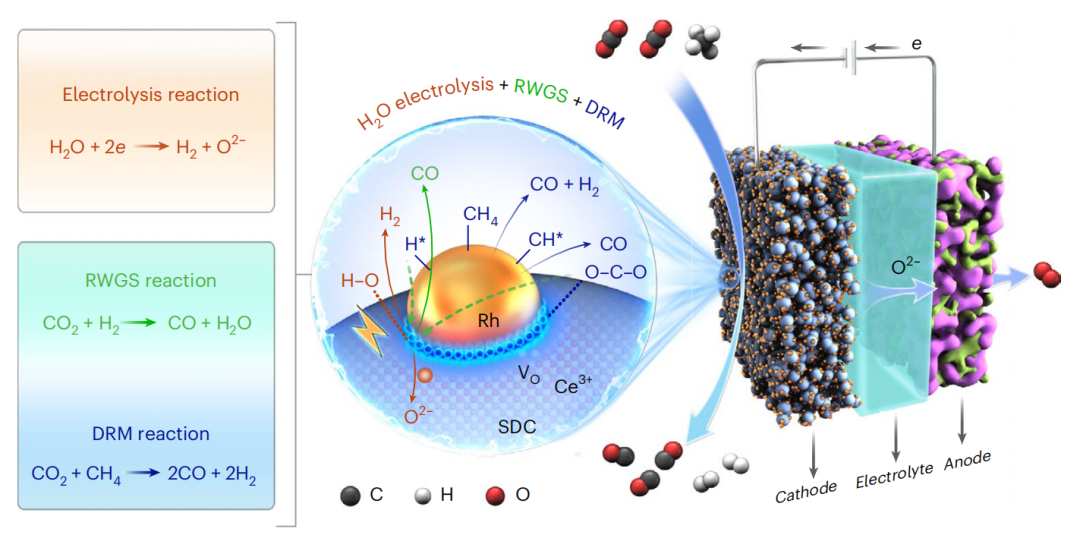
甲烷干重整反应是一种由CO2和CH4生成合成气的反应。虽然该反应通常以1的进料比进行,但设想的未来进料含有更多的二氧化碳,因此需要大量分离以使用所需的CH4。
大连化学物理研究所肖建平研究员、汪国雄研究员、包信和院士等人开发了一种三步串联电-热催化CH4重整反应,用于转化富含CO2的天然气。串联CH4重整工艺与逆水煤气反应与氧离子导电电解膜反应器相结合,其中水电解改变了逆水煤气反应的平衡,促进了合成气的生成,提高了CH4的表观还原性。该催化剂由Rh纳米颗粒在CeO2-x载体上的原位外溶形成,为高催化性能提供了大量的Ce3+-VO-Rhδ+界面活性位点。该串联系统中每个CH4分子使用多达4个CO2分子,提供高CH4和CO2转化率,并产生高选择性的CO和H2。
相关工作以《Super-dry reforming of methane using a tandem electro-thermocatalytic system》为题在《Nature Chemistry》上发表论文。
肖建平,研究员,博士生导师,国家杰出青年科学基金获得者。2019年至今于中科院大连化学物理研究所受聘为研究员,担任计算和数据驱动催化研究组(511组)组长。以通讯/共同通讯作者身份在国际知名刊物上发表论文80余篇。目前主要研究领域为多相催化和电催化的理论计算模拟和实验研究,主要研究的反应包括合成氨、合成氨基酸、氮气活化、烷烃脱氢和芳构化、合成精细化学品等。
汪国雄,复旦大学化学系教授,国家杰出青年科学基金获得者。2010年回国先后任大连化学物理研究所副研究员、研究员。研究兴趣:电催化、碳基分子电化学转化、电化学合成氨、电解水制氢。担任《物理化学学报》编委、《电化学》编委、《Green Energy & Environmental》编委、《催化学报》青年编委、《eScience》青年编委,中国化学会二氧化碳化学专业委员会委员。
本文采用原位溶出限制性Rh纳米粒子(NPs)的铈基电极催化剂(Sm0.17Rh0.03Ce0.8O2-δ, SRhC)作为三步串联工艺的多功能催化剂。Rh NPs在SmxCe1-xO2-δ(SDC)载体上析出,具有良好的中等水平金属-载体相互作用(Rh/SDC),提供了大量的Ce3+-VO-Rhδ+界面活性位点(VO,氧空位),并表现出优异的催化活性和稳定性。理论计算和原位表征研究揭示了Ce3+-VO-Rhδ+界面位在操作条件下转移电子和氧的关键作用。对于高CO2含量天然气的转化,该串联系统提供了一个集成设计,对合成气的选择性接近100%,因此在超过2的任何比率下,CH4的明显还原性都接近其最佳值(图1)。
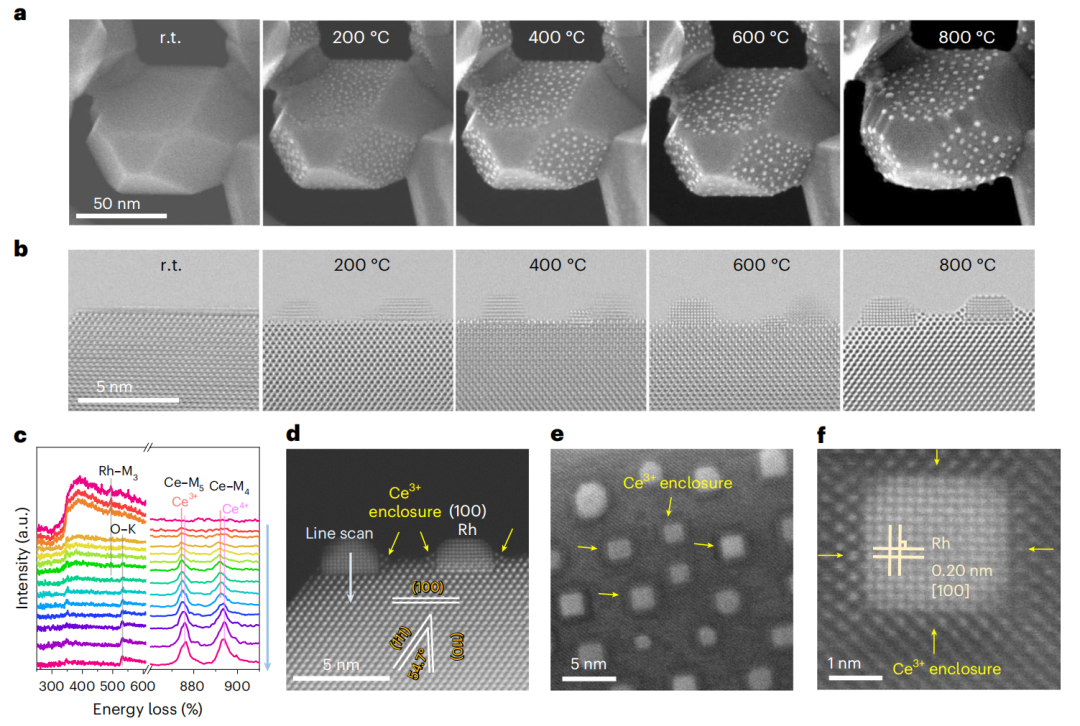
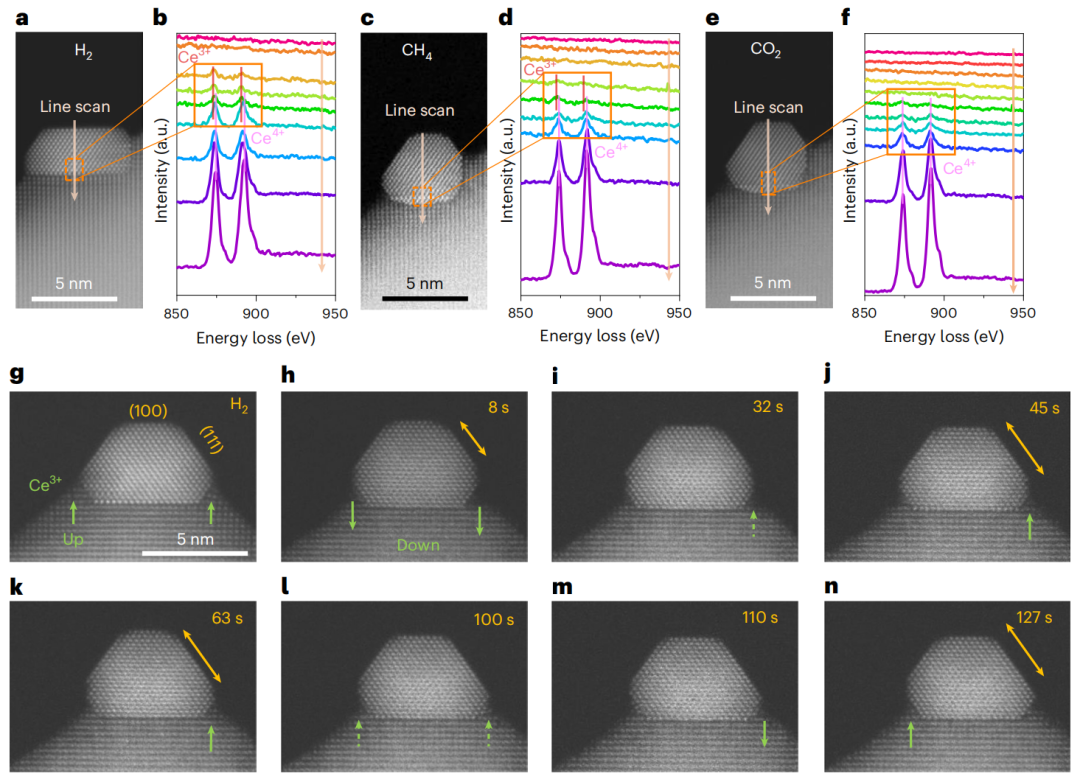
图2 H2作用下原位还原过程中颗粒析出和锚定的监测
本文采用原位像差校正高角度环形暗场扫描透射电子显微镜(HAADF-STEM)对成核和结晶过程进行实时成像(图2),这有望为催化活性的结构特征提供见解。所制备的SRhC具有原始的表面,在SRhC的萤石晶格中Rh离子分布均匀(图2a)。为了监测Rh NPs的原位生长,在室温至800℃的H2条件下获得二次电子STEM图像。SRhC的起始温度较低,为200℃。在连续的析出过程中,观察到Rh NPs在SDC上的析出具有明显的晶面依赖行为。在整个生长过程中,相对于(111)的高指数取向,NPs保持锁定在低指数晶体取向,例如(100)。
在同一位置获得了析出的Rh、铈载体及其界面的“透明”原子分辨率图像,以跟踪成核和结晶过程(图2b)。在整个温度范围内,NP的析出保持各向同性,所有NP与载体表现出相同的形状和相同的取向关系。Rh和SDC载体之间的周长界面用载体上的材料修饰,展示了外溶金属NPs及其与氧化物载体的嵌套外延界面的限制结构方面的形成。
活性Rh/SDC界面在800℃还原环境下保持稳定。图2c、d和补充图5所示的原位STEM和EELS线扫描谱证实了该界面覆盖层为CeO2-x(03+外壳的半封装结构。这种特殊的结构使它们在高温下对粗化具有更强的活性和稳定性。
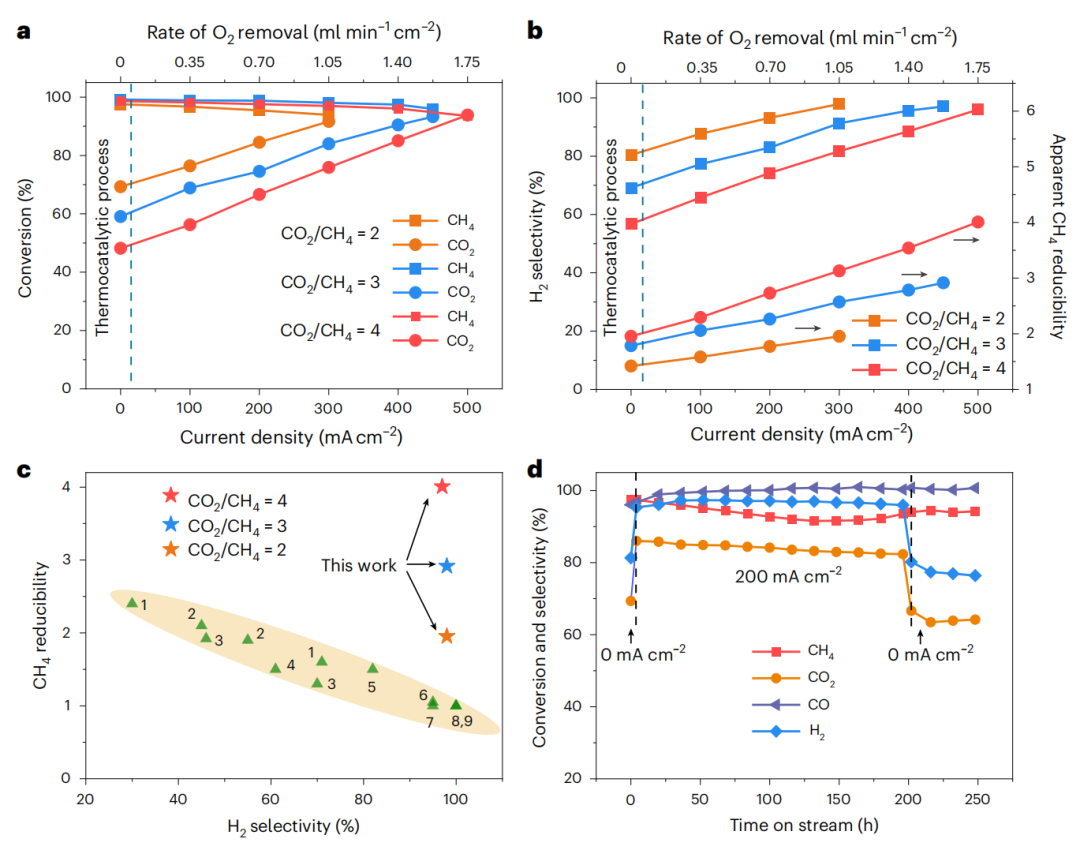
克服热力学限制并从副产物H2O中释放H2的一种有希望的方法是集成EMR进行选择性H2O电解,这将推动反应形成H2和CO。EMR的电解质含有掺杂的LaGaO3钙钛矿,其从H2O中吸收氧气,具有高氧离子电导率。图3a显示了ETC-DRM过程的结果。在EMR系统上施加电流后,随着O2去除率的增加,CO2转化率继续增加。当电流密度分别为300、450和500 mA cm-2,进料比分别为2、3和4时,CO2转化率分别高达91.5%、93.2%和93.9%。在这种情况下,RWGS被认为是排除DRM后过剩CO2转化的主要工艺,而H2O电解被认为是实现高CO2转化率的主要电解工艺(图3b)。CO是唯一的含碳产物,随着O2去除率的提高,H2选择性增强。在最大电流密度下,H2选择性超过96.0%,没有观察到明显的H2O生成(图3b)。
在热催化(TC)-DRM过程中,CH4的还原性在高温下受到限制。重要的是,即使在800℃时,EMR系统的CH4表观还原性也很好,并且随着O2去除率的增加而急剧提高(图3b)。通过该ETC-DRM工艺,CH4的表观还原性显著提高;超过理论最大值3,在任何进料比大于2的情况下都接近理论最大值(图3c)。更重要的是,产物是H2而不是H2O。在200 mA cm-2、800℃、200 h的条件下,测试了Rh/SDC阴极上ETC-DRM反应的耐久性(图3d)。图3d中最显著的结果是,与传统TC-DRM工艺相比,CO2转化率和H2选择性得到了提高,而且没有观察到沉积的碳。在连续操作252h后,Rh NPs仍然锚定在SDC表面,其大小与反应前相同。
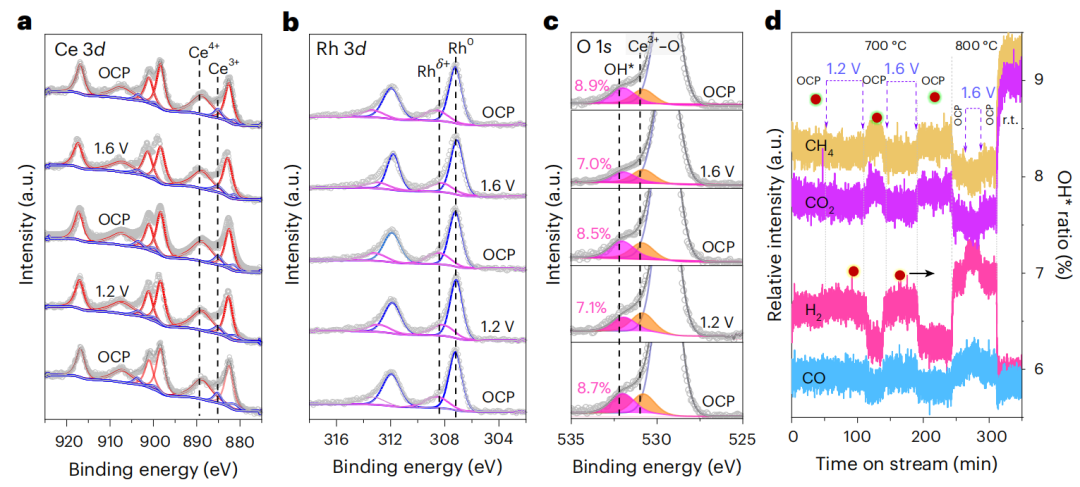
图4 Rh/SDC催化剂在ETC-DRM模式下的NAP-XPS光谱和原位质谱信号
为了研究ETC-DRM模式下催化表面和吸附物的化学性质,在不同的施加电压下获得了原位NAP-XPS光谱(图4)。限制在SDC上的Rh NPs提供了大量具有优异催化性能的Ce3+-VO-Rhδ+界面活性位点。在CO、CO2、CH4和H2同时存在的情况下,Ce3+-VO-Rhδ+界面活性态通过TC-DRM和ETC-DRM工艺在高温下稳定下来(图4a、b)。
当电压应用到电解池上,氧离子泵加到阳极上时,OH基团的峰值减弱(图4c、d)。除了OH基团的消耗外,在线MS信号中还观察到H2和CO的急剧增加(图4d)。回到开路电压后,OH基团的比例增加,H2和CO信号减少。结果表明,在600~800℃的施加电压下,CO和H2的表面OH基团覆盖率和MS信号是动态响应的。这种振荡行为强烈表明,如前所述,施加的电压在吸附的OH基团或H2O分子的电化学还原中起着至关重要的作用,释放H2并将平衡转移到干燥的合成气。
图5 利用原位STEM技术可视化Rh/SDC界面的化学和结构演变
当暴露于反应物中时,金属NPs及其载体经常发生结构和氧化态的变化。为了可视化原子尺度上的动态化学和结构演变,对CH4和CO2反应物存在下的Rh/SDC界面进行了原位STEM研究。H2预处理后,原位STEM图像证实了SDC上受限Rh NPs的溶解(图5a)。原位STEM-EELS线扫描证实,Rh NPs被界面Ce3+包围(图5b中的红色实线)。暴露在H2下的Rh NPs呈现出(100)、(110)和(111)面的形貌。切换到CH4后,NPs逐渐变成球形,并表现出更高比例的(111)面。原位STEM-EELS线扫描结果表明,与Rh NPs密切接触的Ce转变为Ce4+氧化态(图5c、d)。
图5显示了原位暗场STEM图像,该图像监测了Rh NPs和Rh/SDC界面在原子尺度上的结构演变。总的来说,结构和形态的演变归因于吸附物引起的表面能和界面能的变化。此外,本工作还表明,Rh晶面的重构伴随着相关活性位点的优化。Rh NPs在衬底表面的局部位置发生结构波动。此外,在CH4中的Rh NPs倾向于暴露(111)面,并且NPs之间的结构振荡是相同的。Rh(111)面被推断为能量上有利于CH4解离。
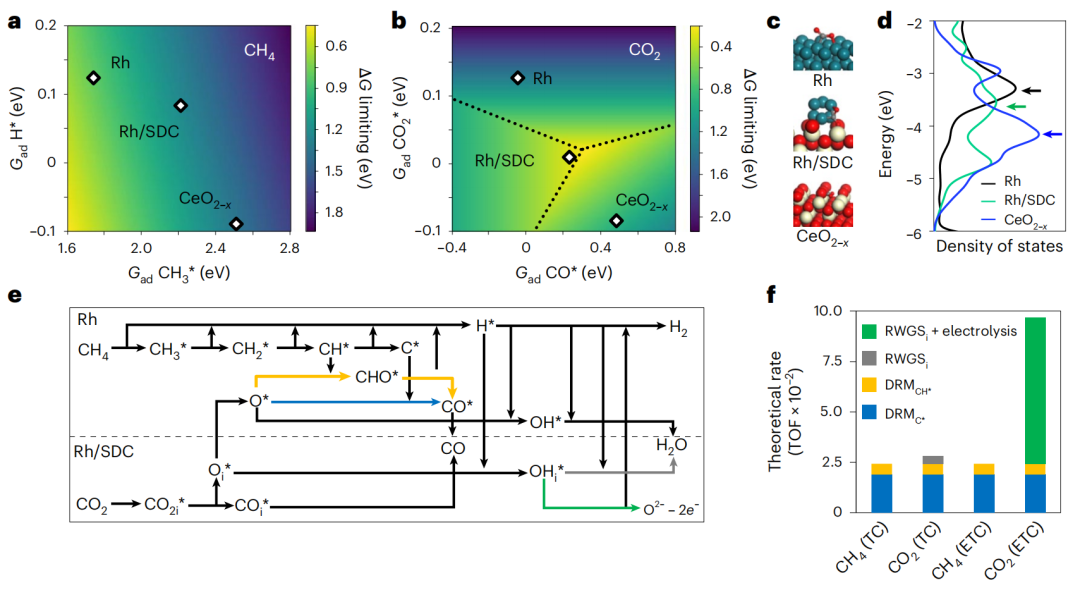
作者进行DFT计算以了解DRM活性。在STEM图像中,选择CeO2-x(100)和Rh(111)表面作为载体面和金属面(即CeO-和Rh)的模型。构建了一种具有外溶Rh簇的CeO2-x(100)结构,研究了在Rh/SDC上Ce3+-VO-Rhδ+界面。为了得到CH4解离和CO2活化的(准)活性趋势,建立了二维反应相图(图6a、b),因为所有吸附自由能(Gad)都与CH3*的Gad和CO2*的Gad有关。
反应相图显示,CH4解离活性依次为CeO2-x4解离的限制步骤的ΔG最小。对于CO2活化,限制步骤是CO2在Rh上吸附,CO2在CeO2-x上解离,而CO在Rh/SDC上解吸。Rh/SDC几乎处于CO2活化的最佳位置,对CO2*具有中等的吸附能力。CO2*在三个表面上的吸附结构和态密度如图6c、d所示。在Rh/SDC和CeO2-x的VO上吸附了CO2*。吸附在Rh/SDC上的CO2*的态密度比Rh低,而电子态密度比CeO2-x少。因此,CO2*在界面位置的吸附强度介于金属表面和载体表面的吸附强度之间,这与DFT计算的能量一致(图6b)。因此,Rh/SDC上的Ce3+-VO-Rhδ+界面位点促进了CO2的吸附和解离,最终提高了总催化性能。
基于实验表征数据和前面描述的热力学分析,Super-DRM的总过程如图6e所示。计算了基本反应(R1-R18)和所涉及的能量。通过C*和CH*消耗O*的DRM进程分别定义为DRMC*和DRMCH*。此外,RWGS发生在界面(RWGSi)和协同过程(RWGSi+电解)。为了探索TC和ETC反应条件下CH4和CO2转化的理论速率(TOF),进行了微动力学模拟(图6f)。两种条件下CH4的TOF均为2.43×10-2 s-1。在TC和ETC反应条件下,CO2的TOFs分别为2.82×10-2 s-1和9.69×10-2 s-1。在TC和ETC反应条件下,迁移的O*在Rh表面与C*和CH*反应,DRM(DRMC*和DRMCH*)过程的贡献率是相等的。
【高端测试,找华算】🏅 同步辐射 全球机时,三代光源,随寄随测!最快一周出结果,保证数据质量!
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!