

在使用Materials Studio(MS)进行材料计算时,电子步不收敛是一个常见的问题,尤其是当你看到“Warning: electronic minimisation did not converge when finding ground state”这样的提示时,是不是感到头疼不已?别担心,今天我们就来聊聊如何解决这个问题!


1. 抛掉Fix Occupancy
首先,在CASTEP Calculation —Electronic-–more-–SCF中找到Fix occupancy,把前面的√去掉,不要勾选,这个方法可以解决90%的电子步不收敛问题。简单操作,效果显著!
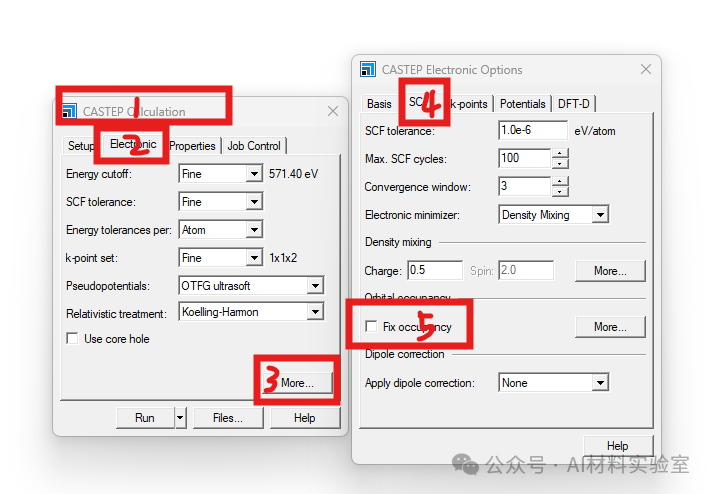
2. 调整Smearing值和Empty Bands
如果问题依然存在,打开Fix occupancy右边的More,可以尝试加大smearing值和empty bands值。这两个参数的调整可以有效帮助系统找到基态。
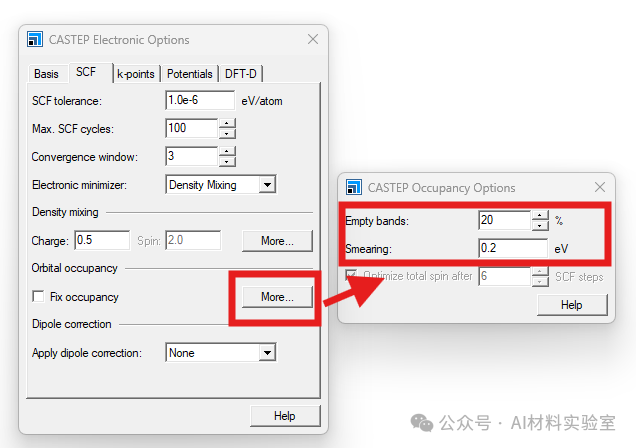
3. 调大Max SCF Cycles
对于较大的体系,建议将Electronic—more–SCF中的Max SCF cycles调大,给系统更多的SCF迭代次数来收敛。
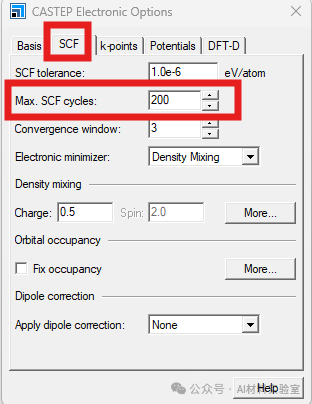
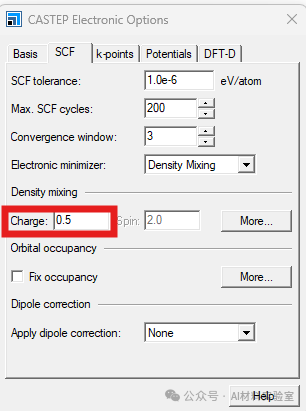
4. 调小Density Mixing中的Charge
如果还是不收敛,可以尝试将density mixing下的charge值调小到0.1-0.2,这有助于系统更快地找到平衡。
5. 处理电子自旋极化
如果体系中存在电子自旋极化,打开后电子步不收敛,可以尝试减小density mixing中的spin值。
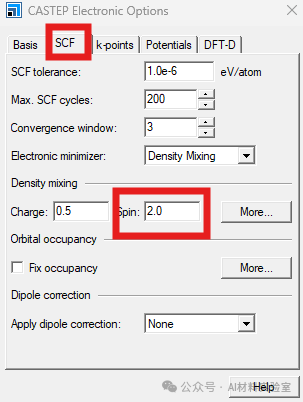
6. 设置原子磁矩
对于有磁性的体系,可以在菜单栏中找到modify,打开其中的electronic configuration界面,在Spin中设置合适的原子初始磁矩。
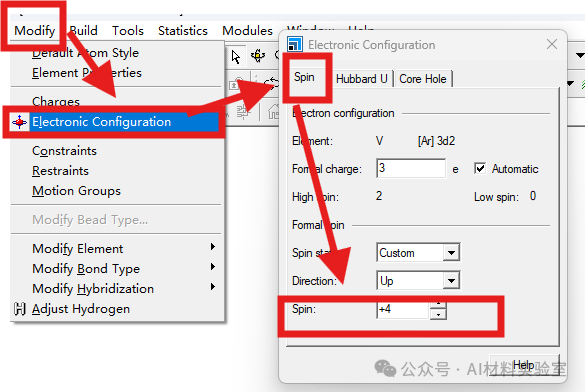
7. 更换Minimizer
如果以上方法都不奏效,可以将SCF选项卡中的minimizer从density mixing换成all bands/EDFT,虽然计算会变慢,但收敛性会大大提高。
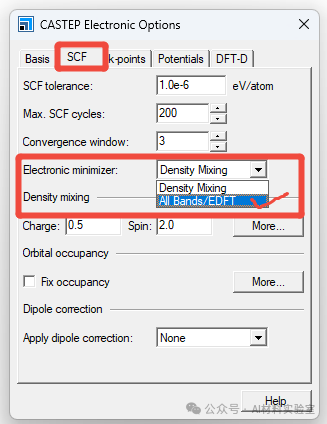
8. 检查K点设置
确保K点设置合理,尤其是对于较大的体系,K点过少可能导致电子步不收敛。
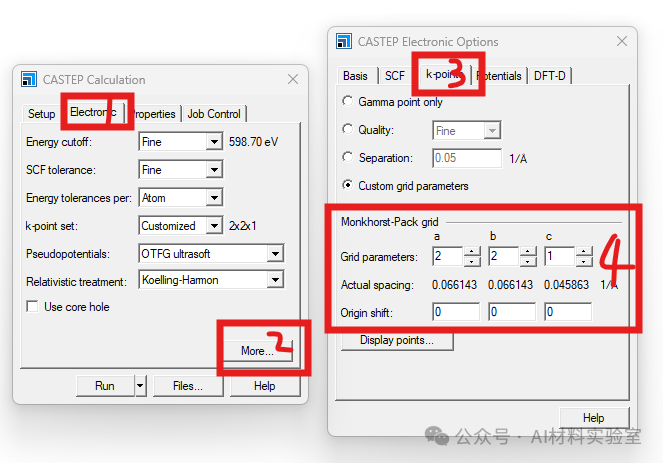
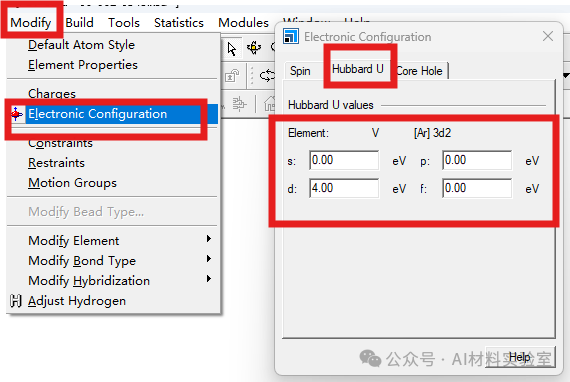
9. 使用DFT+U方法
对于强关联体系,可以尝试使用DFT+U方法,在菜单栏中找到modify,打开其中的electronic configuration界面,在Hubbard U中设置合适的 U值,这有助于更好地描述电子间的相互作用,从而提高收敛性。

总结
电子步不收敛是MS计算中的常见问题,但通过合理的参数调整和方法选择,大多数问题都可以得到解决。希望这些技巧能帮助你在材料计算中更加得心应手!如果你有更多问题,欢迎留言讨论,或者参加我们的MS系列课程,深入了解更多高级技巧!
找华算做计算👍专业靠谱省心又省时!
益于理论计算化学的快速发展,计算模拟在纳米材料研究中的运用日益广泛而深入。科研领域已经逐步形成了“精准制备-理论模拟-先进表征”的研究模式,而正是这种实验和计算模拟的联合佐证,更加增添了论文的可靠性和严谨性,往往能够得到更广泛的认可。
华算科技已向国内外1000多家高校/科研单位提供了超过50000项理论计算和测试表征服务,部分计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。
