

在使用Materials Studio(MS)进行DMol3结构优化时,你是否遇到过这样的问题:明明设置了三个收敛标准,为什么只有两个收敛了,任务就成功结束了?
今天,我们就来详细解答这个问题,并分享如何正确设置收敛标准,确保优化任务按预期完成。
问题描述
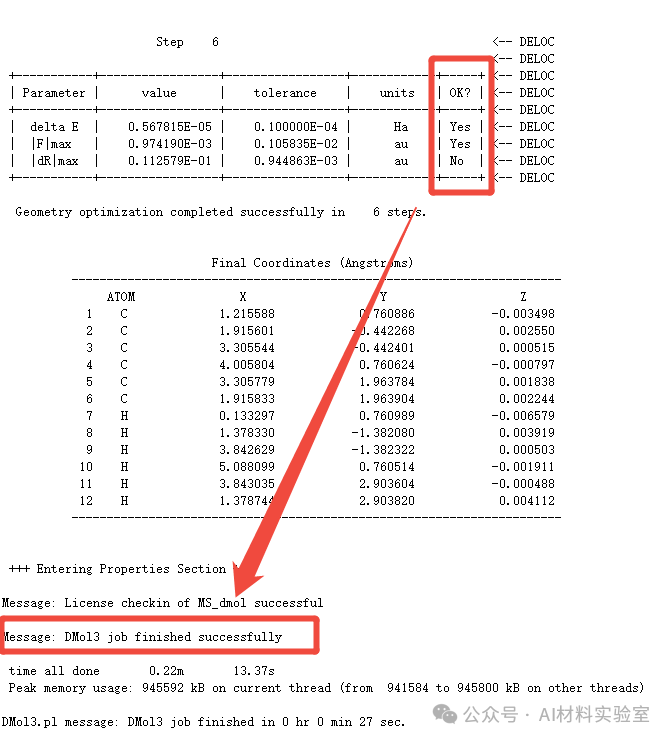
在DMol3结构优化任务中,通常有三个收敛标准:能量变化(delta E)、最大力(|F|max)和最大位移(|dR|max)。然而,很多用户发现,即使只有两个标准(如delta E和|F|max)显示为“Yes”,任务也会成功结束,而|dR|max仍然显示为“No”。这是为什么呢?


原因解析
从MS的help中可以查询到,在DMol3计算中,默认情况下,Opt_Converge_All参数是关闭的(off)。这意味着,能量、力和位移三者中只要有两个满足收敛判据,任务就会成功结束。如果你希望三者都满足收敛判据,必须手动将Opt_Converge_All设置为on。


如何设置修改设置?
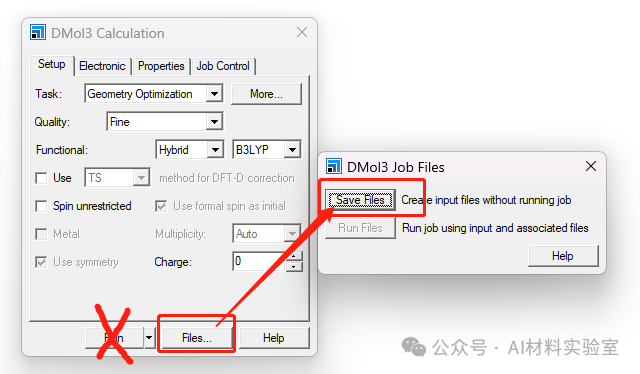
1. 保存计算参数
在设置好所有计算参数后,在DMol3 Calculation界面,不要直接点击“Run”,而是点击旁边的“Files”,然后选择“Save Files”。这时会生成一个包含输入文件的目录。
2. 修改input文件
打开生成的input文件,你会发现其中没有Opt_Converge_All参数。需要手动添加一行:
Opt_Converge_All on
这样,系统会强制要求能量、力和位移三者都满足收敛判据。

3. 保存修改
修改好input文件,在菜单栏找到保存按钮,点击保存。
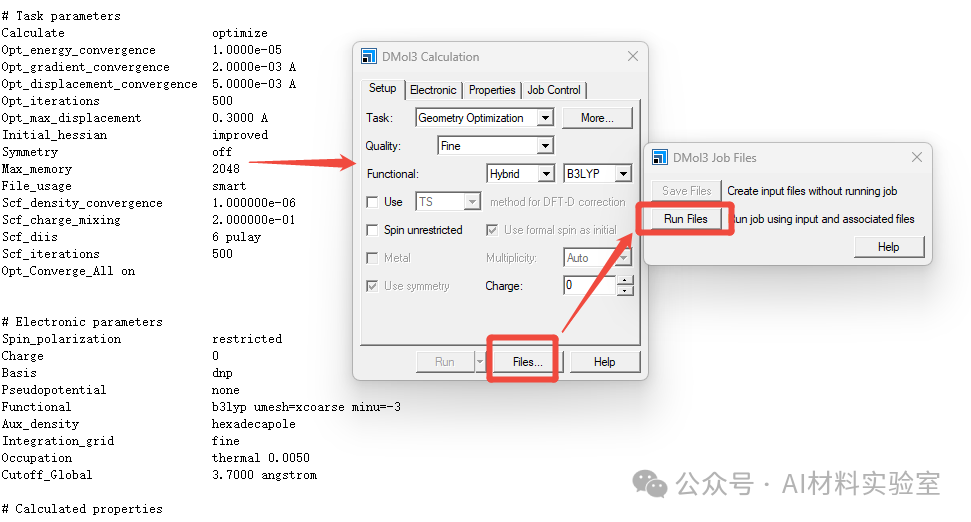
4. 提交任务
保存input文件后,不要退出input文件界面,还是点击DMol3 Caculation中的Files,然后选择“Run Files”提交任务。
在这4步中重点需要注意事项:
· 不要直接点击“Run”:修改input文件后,务必通过“Run Files”提交任务,否则修改无效。
· 确保input文件正确:需要在input界面点击“Run Files”,否则该选项可能会显示为灰色,无法提交。

总结
通过正确设置Opt_Converge_All参数,你可以确保DMol3结构优化任务严格按照能量、力和位移三个收敛标准执行。这一技巧不仅提高了计算的准确性,还能避免因收敛标准不满足而导致的结果偏差。
找华算做计算👍专业靠谱省心又省时!
益于理论计算化学的快速发展,计算模拟在纳米材料研究中的运用日益广泛而深入。科研领域已经逐步形成了“精准制备-理论模拟-先进表征”的研究模式,而正是这种实验和计算模拟的联合佐证,更加增添了论文的可靠性和严谨性,往往能够得到更广泛的认可。
华算科技已向国内外1000多家高校/科研单位提供了超过50000项理论计算和测试表征服务,部分计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。