

理论计算在电池领域扮演着至关重要的角色,它不仅为电池材料的设计和优化提供了强有力的理论支持,还极大地推动了电池性能的提升和新型电池技术的发展。通过精确的计算模拟,研究人员能够深入理解电池材料的结构特性、电化学行为以及反应机理,从而在材料选择、性能预测和安全性评估等方面实现更加精准和高效的决策。
一、吸附能
通过吸附能计算,可以确定电解质中哪些离子或分子易于在电极表面吸附,这对于选择或设计合适的电解质非常重要。
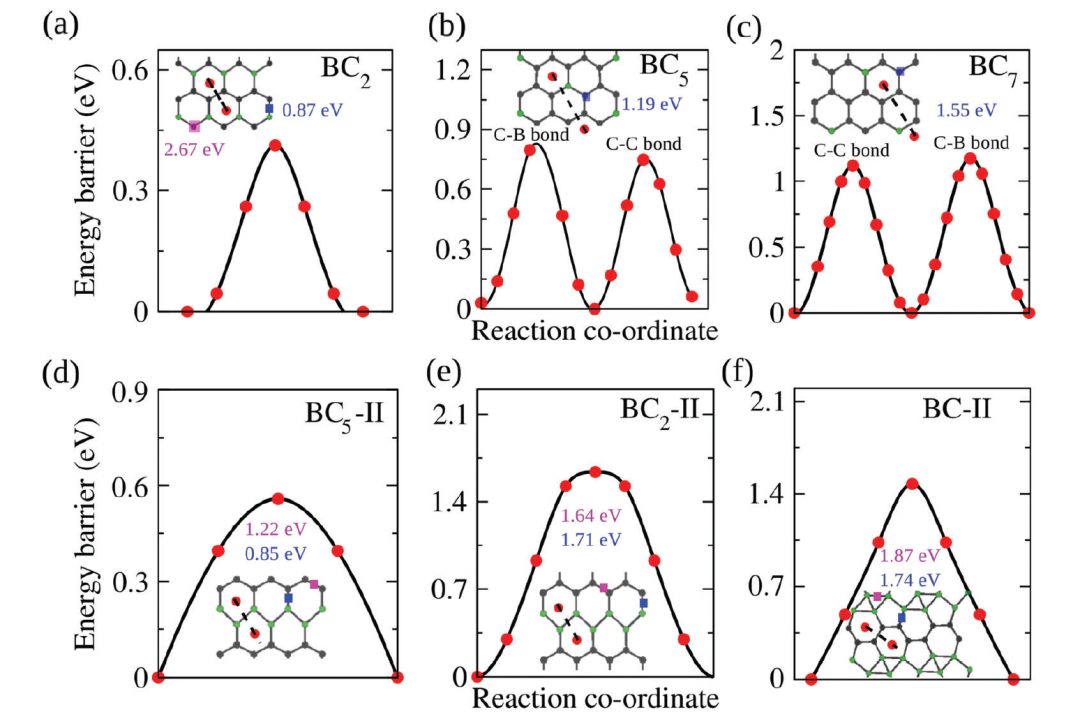
二、离子迁移势垒
通过离子迁移势垒可以评估离子在电解质中的传输效率,这对于优化电池设计、提高电池性能提供了重要的理论支持和指导。
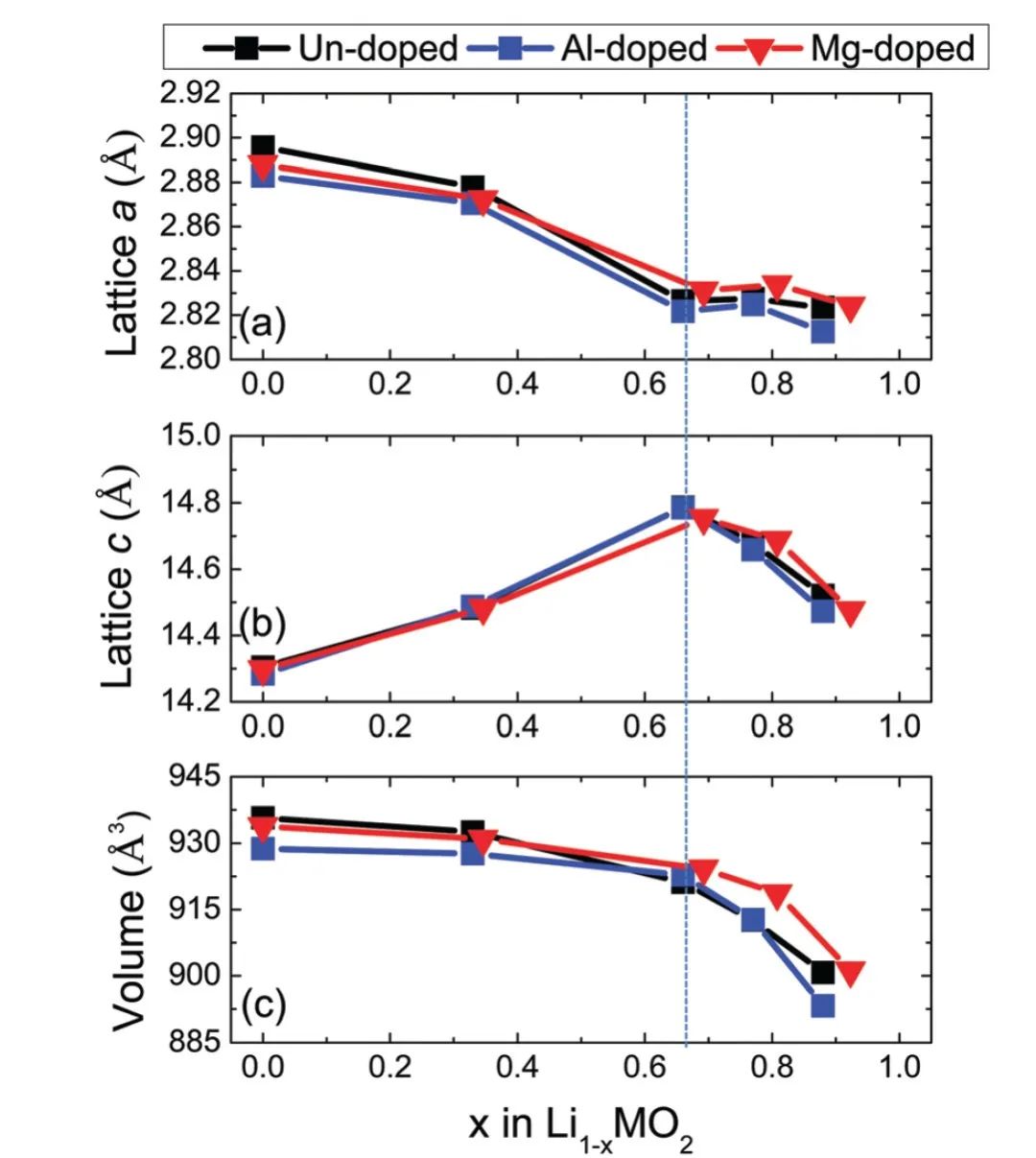
三、材料改性模拟
引入特定的掺杂、缺陷等,会影响电极材料的电子结构、晶格结构、离子电导率、理论容量、电压曲线等,有助于开发高效率的新型电池。
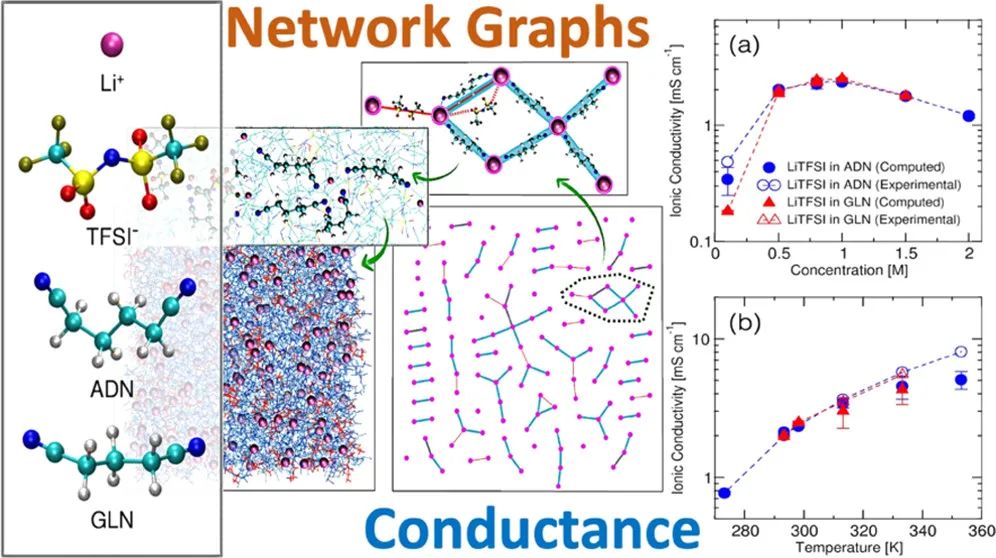
四、分子动力学
MD模拟有助于理解离子吸附、离子迁移、溶剂化壳层的形成和分解过程,以及在充放电中的晶格膨胀、收缩和相变等。
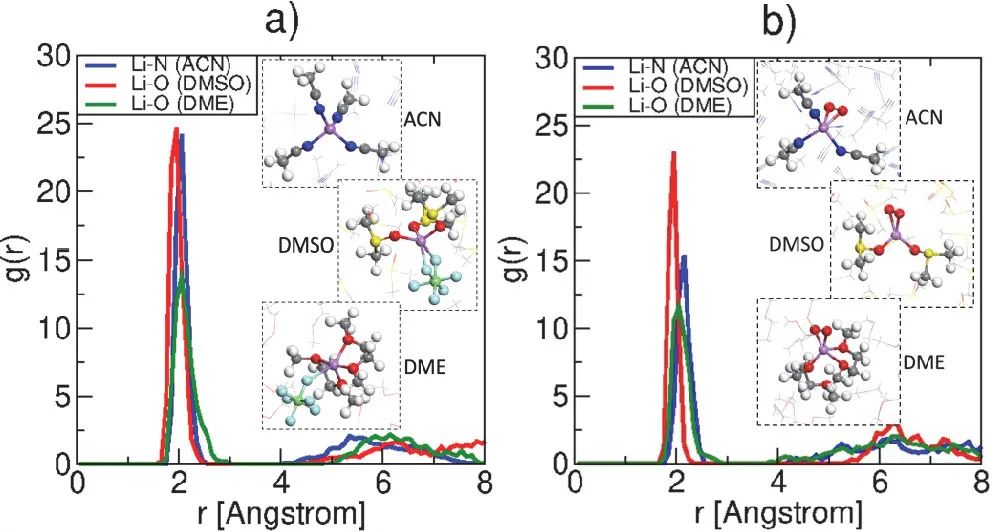
五、溶剂化结构
溶剂化结构决定了离子在电解质中的传输方式和效率。通过计算,可以揭示离子与溶剂分子之间的相互作用,从而优化离子传输过程。
六、界面反应模拟
通过研究电解液/电极的界面相互作用、金属锂枝晶的生长和抑制机理、特定晶面的反应活性等,有助于设计高性能电池。
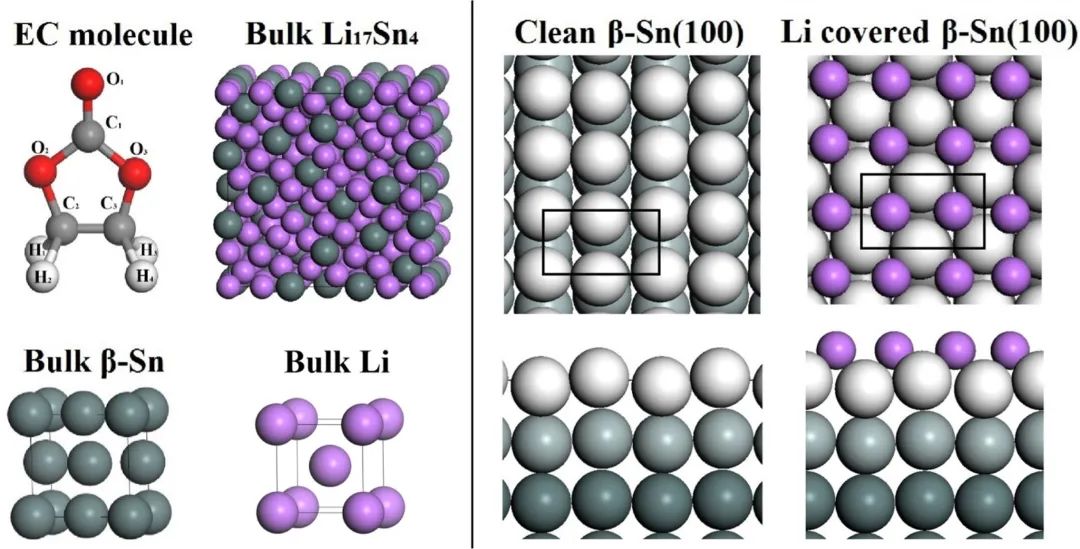
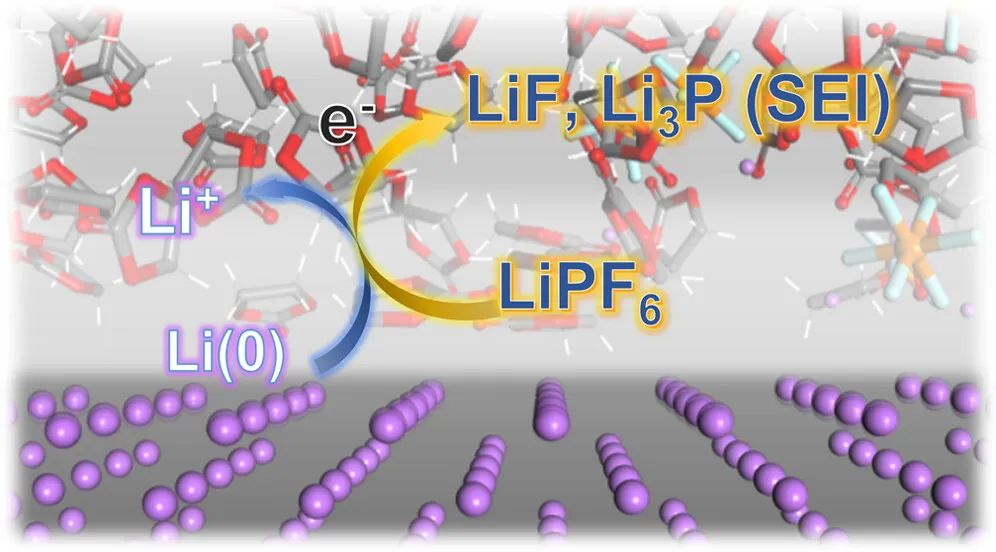
七、SEI膜机理模拟
研究SEI膜的形成机制、成分和结构,对于深入理解电池工作原理、提高电池性能和开发新型电池材料都具有重要的理论和实际意义。
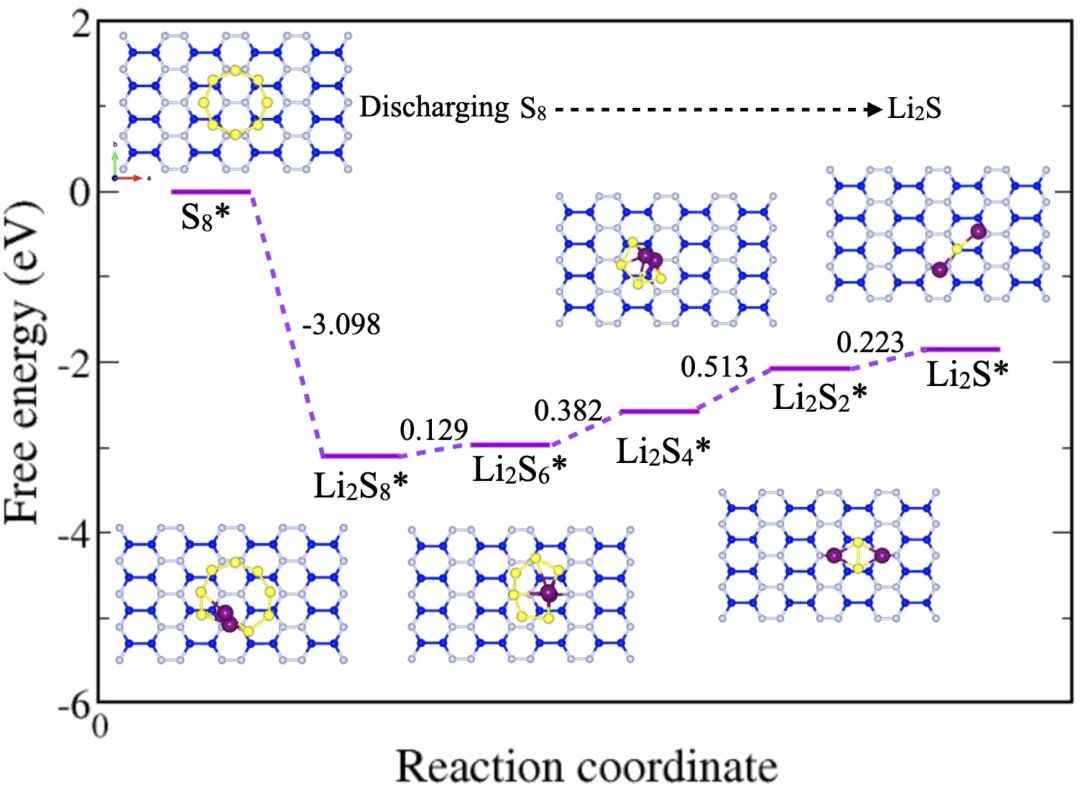
八、硫还原反应(SRR)
硫还原反应是锂硫电池中放电过程的核心步骤,有助于揭示硫分子在电极表面的还原过程、中间产物及其转化路径,从而深入理解电池的工作机理。
找华算做计算👍专业靠谱省心又省时!
益于理论计算化学的快速发展,计算模拟在纳米材料研究中的运用日益广泛而深入。科研领域已经逐步形成了“精准制备-理论模拟-先进表征”的研究模式,而正是这种实验和计算模拟的联合佐证,更加增添了论文的可靠性和严谨性,往往能够得到更广泛的认可。
华算科技已向国内外1000多家高校/科研单位提供了超过50000项理论计算和测试表征服务,部分计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。