
目的:演示如何使用Blends模块筛选分子相互作用。
本教程包括如下部分:
-
开始 -
建立输入结构模型 -
设置Blends模块计算参数并运行计算 -
结果分析
注意:为了确保您可以完全按照预期的方式学习本教程,您应该使用“设置管理器(Settings Organizer)”对话框确保项目中所有参数都设置为BIOVIA的默认值。

 打开“导入文档Import Document”对话框。导航到路径文件夹Structures/repeat units/oxides,并选择oxyethylene.xsd文件,单击打开按钮。
打开“导入文档Import Document”对话框。导航到路径文件夹Structures/repeat units/oxides,并选择oxyethylene.xsd文件,单击打开按钮。本项目引入了氧乙烯结构。结构中的头原子和尾原子分别由原子周围的青色和红色笼状标志示出。
重复上述步骤,将Structures/repeat-units/olefins/propylene.xsd和Structures/repeat-units/acrylates/acrylic_acid.xsd导入项目。
2、建立输入结构模型
在Blends模块计算中,可以筛选聚合物之间、聚合物-溶剂或溶剂-溶剂的相互作用。可以通过指定重复单元的头原子和尾原子来定义聚合物。
作为建模过程的一部分,在Blends模块中提交重复单元进行计算之前,应优化重复单元的几何构型。这需要用到Forcite模块。
使oxyethylene.xsd成为当前活动文档。从菜单栏中选择Modules | Forcite | Calculation以打开Forcite Calculation对话框。
在Setup选项卡上,将任务Task更改为“几何优化Geometry Optimization”。
在Energy选项卡上,从力场Forcefield下拉列表中选择Dreiding,并将电荷Charges更改为使用QEq电荷Charge using QEq。单击Run按钮。
-
oxyethylene.xsd:由初始结构优化得到的几何构型的3D原子文档。
-
oxyethylene – Calculation:Forcite计算任务参数设置的.xml文件。单击此文件将打开Forcite Calculation对话框,其中包含计算设置的参数。
-
oxyethylene Convergence.xcd:能量变化和梯度法线变化曲线图的图表文件。
-
oxyethylene Energies.xcd:焓变曲线图的图表文件。
-
Status.txt:实时计算状态更新的文本文件。
-
oxyethylene.txt:初始计算任务参数设置以及初始和最终结构的能量分量的文件。
分别对acrylic_acid.xsd和propylene.xsd两个结构重复上述的几何优化计算。关闭Forcite Calculation对话框。
 ,然后从下拉列表中选择Calculation。
,然后从下拉列表中选择Calculation。
Blends Calculation对话框的Setup选项卡
-
Mixing混合:执行结合能和配位数计算。预测混合能、相互作用能和chi参数值。 -
Binding energies结合能:仅计算结合能。可以根据相互作用能进行快速筛选。 -
Coordination numbers配位数:计算一对分子的配位数。
-
Input:输入结构的文件夹。 -
Lowest energies:3D轨迹文件的文件夹,其中保存每组base和screen角色的组合的最低能量构型。对于每个这样的组合,Blends返回三个轨迹文件,其中包含base-base、base-screen和screen-screen三种组合对的构型。将忽略任何与现有组合重复的组合对。 -
oxyethylene – Calculation:Blends计算任务参数设置的.xml文件。单击此文件将打开Blends Calculation对话框,其中包含您为计算设置的参数。 -
oxyethylene.txt:计算任务运行信息和警告信息的文本文档。 -
Configurations.xsd:当前正在采样的结构的3D原子文档。随着计算任务的进行,此文件将实时更新。 -
Energies.xcd:当前正在采样的结构能量变化曲线的图表文档。随着计算任务的进行,此文件将实时更新。 -
Status.txt:显示运行状态的文本文档。随着计算任务的进行,此文件将实时更新。 -
oxyethylene.std:Blends模块运行结果的数据表。 -
4、结果分析
-
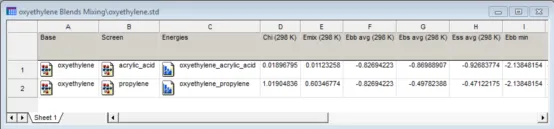
数据表文件oxyethylene.std为对筛选结果的总结,每行代表一对结构组合的计算。 -
注:由于Blends模块计算的特性,结果会有所波动。因此,计算得到的结果完全可能与下面显示的结果不完全匹配。 -
-
Blends计算输出的数据表 -
A列和B列为筛选计算中使用的角色为role和screen分子的结构。列C为一个图表文档,其中包含base-base、base-screen和screen-screen组合的相互作用能曲线图。D列和E列包含chi参数和混合能量的预测值。列F-Q包含相互作用能量的各个分项,并给出了平均值、最小值和最大值。列R-U包含每个角色为role和screen分子组合的配位数。 -
双击单元格B2打开丙烯单体的数据表详细视图Study Table Detail View。 -
头部和尾部原子被标记为名为NONCONTACT的非接触基团。 -
关闭详细视图。 -
数据表中的能量图包含role和screen分子组合的结合能分布。 -
双击单元格C1和C2中的图表。 -
两个图表显示了base-base (Ebb)、base-screen (Ebs)和screen-screen (Ess)的结合能分布曲线图。 -
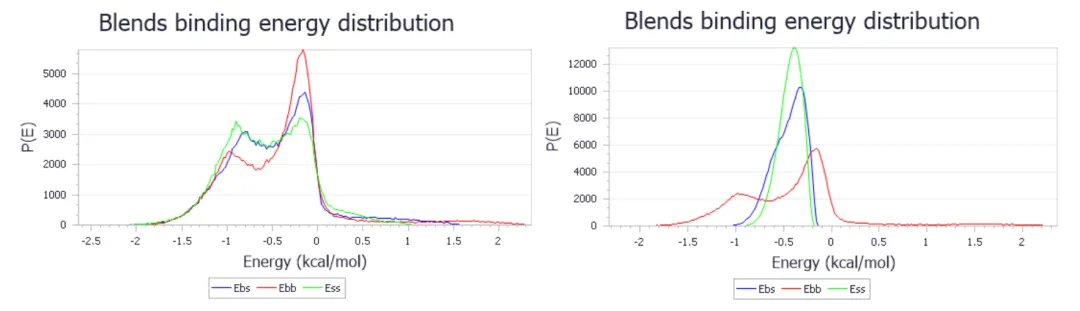
-
两个base-screen组合的结合能分布:氧乙烯-丙烯酸(左)和氧乙烯-丙烯(右) -
两个曲线图中的base-base结合能看起来应该相似,因为它们在两种情况下都对应于氧乙烯-氧乙烯组合。然而,screen-screen结合能差异明显。对于丙烯酸的计算结果更类似于氧乙烯。这是一个很好的指标,表明结构将容易混溶。相比之下,丙烯相互作用差距较大,表明混溶性差。 -
关闭两个详细视图。 -
关于混溶性的下一个指标是χ和Emix值。 -
检查D列和E列中的值。 -
χ (chi)值、Emix值接近于零表示混溶性好。χ和Emix值越高,组合对的混溶性越差。这些值验证了从能量分布图得出的结论,即聚氧乙烯将易于与聚丙烯酸混溶,但与聚丙烯较难混溶。 -
除了得到这些初步结果外,Blends还提供了进一步分析的工具。通过选择数据表中所要研究的的聚合物组合对应的行,并从Blends Analysis对话框中选择分析任务,可以进行结果分析。 -
在数据表中,选择第1行。从菜单栏中选择Modules | Blends | Analysis以打开Blends Analysis对话框。选择默认分析模式,即chi parameter后,单击Analyze按钮。 -
将得到一个图表文档Chi parameter.xcd,显示混合有聚丙烯酸的聚氧乙烯χ值与温度的关系。还可以一次对多个数据表行进行分析。 -
关闭Chi parameter.xcd,并在提示是否将文件保存在工程的中时单击No按钮。 -
按住SHIFT键,单击数据表中的第2行。现在应选择了数据表中的两行。在Blends Analysis对话框中,单击Analyze按钮。 -
这一次,曲线图显示了两个base-screen组合χ值与温度的关系。随着温度升高,这两个图中的值应接近0,但在低温下有所不同。 -
对于聚合度N相同的聚合物混合物,存在一个临界值χcrit=2/N。在χ高于临界值的温度下,混合物将发生相分离。在相图中,两相中的浓度位于双结点曲线上。您可以使用相图分析获得双结点曲线。计算N=25的相图,使χcrit=0.08。 -
在Blends Analysis对话框中选择“相图Phase diagram”,并将角色为Base和Screen分子的聚合度Degree of polymerization更改为25。单击Analyze按钮。 -
氧乙烯-丙烯的相图应包含一个临界点,其温度为χ=χcrit=0.08。低于此温度时,体系发生相分离,其浓度由双结点曲线(binodal线)给出。如果浓度和温度在亚稳均相极限线(spinodal线)范围内,相分离是自发的。在双结点曲线和亚稳均相极限线之间,系统是亚稳的;只有远离平衡的过程才能在这种情况下实现相分离。 -
氧乙烯-丙烯酸的临界点要么不存在,要么比氧乙烯丙烯小得多,表明组分在任何成分下都是可混溶的。如果对于任何温度,χ(T)曲线均低于0.08,则不存在临界点,因此也不存在双结点曲线和亚稳均相极限线。如果曲线两次穿过χcrit,相图在两相区的两侧有两个临界点。 -
在本教程的最后一部分中,将研究能量最低的构型轨迹,可以仅播放轨迹。 -
双击文件夹Lowest energies中的oxyethylene acrylic_acid.xtd文件。从菜单栏中选择View | Toolbars | Animation。单击Animation工具栏上的Play按钮  。
。 -
轨迹中的帧将以很快的速度播放。另一种方法是使用Forcite Analysis对话框来分析轨迹。 -
在Modules工具栏上,单击Forcite按钮  并从下拉列表中选择Analysis以打开Forcite Analysis对话框。选择“在数据表中查看View in a study table”,然后选中“包含结构Include structures”复选框。单击View按钮。
并从下拉列表中选择Analysis以打开Forcite Analysis对话框。选择“在数据表中查看View in a study table”,然后选中“包含结构Include structures”复选框。单击View按钮。 -
轨迹以及相关的能量数据将显示在数据表文档中。可以使用数据表工具按能量分项而不是总能量进行排序。 -
选择E列,van der Waals energy范德华能量。单击Study Table工具栏上的“升序排序Sort Ascending”按钮。 -
数据表中的行将从顶部开始按van der Waals能量最低的构象排列。 -
还可以从数据表中提取结构,并将其覆盖在三维原子集合文档中。 -
选择A列,在其中一个选定单元格中单击鼠标右键,然后从快捷菜单中选择“提取到集合Extract To Collection”。将出现一个警告对话框,通知此操作可能需要一些时间。单击OK按钮。 -
将产生一个新的3D原子集合文件Extracted From oxyethylene acrylic_acid.xod,可以轻松移除NONCONTACT标签。 -
在集合文件中单击鼠标右键,然后从快捷菜单中选择“标签Label”以打开Label对话框。单击Remove All按钮并关闭Label对话框。 -
在集合文档中,您应该能够沿着分子的一半找出三个化学键集中的区域。 -
重复上述步骤,将oxyethylene propylene.xtd提取为集合文件进行查看。 -
该例中,有两个主要的相互作用位点,它们不像聚氧乙烯-聚丙烯酸组合那样分散在分子周围。 -
本教程到此结束。