

 一、什么是单原子体系
一、什么是单原子体系 单原子体系是指将孤立的单个金属或非金属原子(如 Fe、Co、Pt、N、S 等)嵌入到基底材料(如石墨烯、氧化物、硫化物、氮化碳等)的晶格或表面缺陷位点中,形成原子级分散的活性位点。这种结构既不同于传统的纳米颗粒催化剂,也不同于均匀的本征材料,而是兼具原子级精确调控和基底材料稳定性的独特体系。
主要特点:
原子级分散:掺杂的单个原子均匀分布在基底上,无团聚现象,可实现接近100%的原子利用率。
独特的电子结构:单原子与基底之间的强相互作用(如配位键、电荷转移)会显著改变局域电子态,影响催化活性。
明确的活性位点:由于活性中心为孤立的单原子,其催化机制更易通过理论计算(如DFT)和实验手段(如XAS、STEM)精确解析。
可调控的配位环境:通过改变基底(如石墨烯、MOFs、g-C₃N₄)或掺杂位点(如空位、边缘位、杂原子配位),可优化催化性能。
二、DFT计算在单原子体系中的使用:
1. 单原子掺杂体系的结构稳定性
计算掺杂体系的形成能是评估材料掺杂稳定性的重要方法。若形成能为负值,表明掺杂过程在热力学上是自发进行的,掺杂后的结构比原始体系更稳定。这种稳定性源于掺杂原子与基体材料之间的化学键合作用,使得体系总能量降低。
形成能的大小还能反映掺杂的难易程度,掺杂形成能越小说明掺杂越容易发生,这为材料设计和性能优化提供了重要理论依据。
DOI: 10.6023/A20060223
2. 电子结构的调控机制
在单原子体系中,掺杂是调节材料电子结构和导电性的有效手段。通过引入不同价态的掺杂原子,可以改变体系的电荷分布,从而调控其带隙宽度和导电类型(n型或p型)。
例如,在石墨烯中掺杂氮(N)或硼(B)原子,会引入额外的电子或空穴,使其带隙打开或形成杂质能级,从而增强导电性。类似地,在单层过渡金属硫族化合物(如MoS₂)中掺杂金属或非金属原子,可以调节其直接/间接带隙,优化光电子性能。
此外,单原子掺杂还能引入局域态,影响载流子迁移率,进而调控材料的电输运特性。这些调控机制在半导体器件、催化及能源存储等领域具有重要应用价值。
DOI: 10.1016/j.jcou.2024.102988
3. 电荷分布与极化
在单原子催化体系中,掺杂是调节电荷分布与极化的有效策略。通过引入异质原子(如N、S、P等非金属或过渡金属原子),可以改变宿主材料的电子结构,诱导电荷重新分布。这种电荷调控能够增强活性位点的局域极化,优化中间产物的吸附能,从而提升催化性能。
例如,在石墨烯或MXene等二维材料中掺杂杂原子,可打破电荷对称性,形成电子富集或缺电子区域,促进反应物的活化。此外,掺杂还能调节费米能级位置,影响材料的氧化还原特性。
DOI: 10.1016/j.ijhydene.2024.05.124
4. 催化活性与反应机理的验证
在单原子催化体系中, DFT模拟可构建可能的反应路径,计算各基元反应的能垒,揭示决速步骤及关键中间态,为反应机理提供理论验证。结合过渡态计算(TS),还能预测反应速率和选择性。通过与实验表征(如XAS、FTIR等)对比,DFT计算能有效验证单原子催化剂的活性起源及反应机制,为理性设计高效催化剂提供理论指导。
DOI: 10.1016/j.cattod.2024.114560
5. 缺陷与掺杂协同作用
在单原子体系中,缺陷与掺杂的协同作用可以显著调控材料的电子结构、催化活性和稳定性。缺陷(如空位、间隙原子或位错)能够提供额外的活性位点,而掺杂(如引入异质原子)可以进一步调节局域电子态密度和电荷分布。
二者的协同效应可能增强催化性能,例如在单原子催化剂(SACs)中,金属原子锚定在缺陷位点(如碳基材料中的氮空位或石墨烯中的拓扑缺陷)可优化金属-载体相互作用,提高催化活性和选择性。此外,缺陷与掺杂的协同还可能影响电荷转移、能带结构以及材料的稳定性,为设计高效功能材料提供新的调控策略。
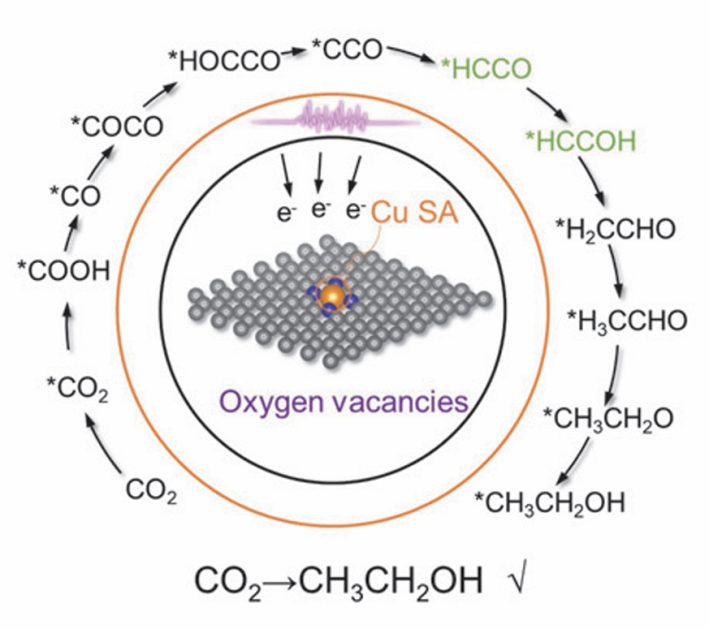
DOI: 10.1016/S1872-2067(23)64587-5
找华算做计算👍专业靠谱省心又省时!
益于理论计算化学的快速发展,计算模拟在纳米材料研究中的运用日益广泛而深入。科研领域已经逐步形成了“精准制备-理论模拟-先进表征”的研究模式,而正是这种实验和计算模拟的联合佐证,更加增添了论文的可靠性和严谨性,往往能够得到更广泛的认可。
华算科技已向国内外1000多家高校/科研单位提供了超过50000项理论计算和测试表征服务,部分计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。
