



超低温锂金属电池面临诸多重大挑战,包括离子传输迟缓以及在高功率条件下锂枝晶不受控制地形成。理想的电解质需要具备高载流离子浓度、低粘度、快速去溶剂化以及稳定的界面等特性,但要平衡这些属性仍是一项艰巨的任务。
清华大学刘凯、Zhang Weili等人设计并合成了一种多功能添加剂——全氟烷基磺酰基季铵硝酸盐(PQA-NO3),它兼具阳离子(PQA+)和阴离子(NO3–)成分。PQA+与锂金属原位反应,形成富含无机物的固态电解质界面(SEI)膜,从而增强锂离子通过该膜的传输。NO3–则形成富含阴离子、溶剂含量低的溶剂化结构,提高正极/电解质界面的氧化稳定性,并减少锂离子与溶剂之间的相互作用。这使得基于乙醚的电解质能够实现高电压耐受性、提高离子电导率以及降低去溶剂化能垒。采用所开发电解质的Li(40微米)||NMC811(3 mAh/cm2)纽扣电池在 -60℃下表现出稳定的循环性能,而450 Wh/kg的软包电池在-85℃时仍能保持48.1%的容量,实现了 171.8 Wh/kg的比能量。此外,该软包电池在-50℃下的放电速率为 3.0 C,达到了938.5 W/kg的比功率,表明了其出色的低温性能。
相关工作以《Multifunctional electrolyte additive for high power lithium metal batteries at ultra-low temperatures》为题在《Nature Communications》上发表论文。
刘凯,清华大学副教授,特别研究员,博士生导师,国家高层次青年人才。长期从事新能源高分子膜材料(如固态锂电池电解质膜,碱性离子膜)和电池安全材料研究。2009-2014年博士毕业清华大学化学系高分子化学与物理专业,导师:张希院士。2014-2019年于斯坦福大学材料科学与工程系从事博士后研究工作,合作导师:崔屹教授。2019入职清华大学化学工程系。曾获《麻省理工科技评论》“35岁以下科技创新35人”(TR35)中国榜单,瑞士Dinitris N. Chorafas青年研究奖(全球每年遴选30人)、清华大学学术新秀等多项奖励。
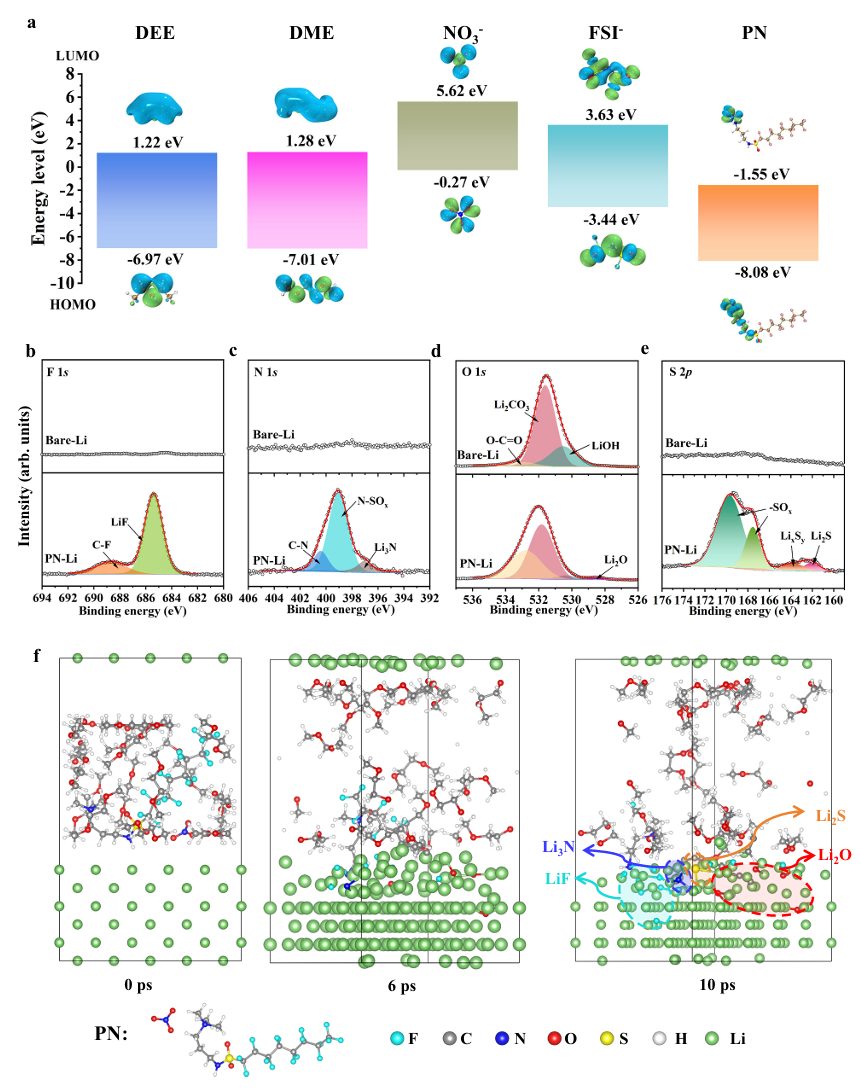
图1 PN添加剂与锂金属之间原位反应的模拟与表征
根据分子前沿轨道理论,具有较低最低未占分子轨道(LUMO)能级的电解质成分在热力学上更倾向于发生还原分解。图1a计算了电解质中不同成分的分子轨道能量水平,结果表明 PN 添加剂分子具有较低的LUMO能级,从而导致其在锂金属表面发生优先分解,并参与SEI形成过程。此外,将锂金属浸入含有0.1 M PN的DME溶液中2小时,然后使用 XPS测试其表面成分。商业锂金属箔通常具有富含Li2CO3和LiOH的天然钝化层(图1b-e),这会增加电极的阻抗和过电位,并且还会对电极表面后续形成的SEI的构建产生影响。然而,经过浸泡处理后,锂金属表面出现了大量诸如LiF、Li2CO3、Li2O、Li2S和Li3N等无机化合物,这表明PN能够与锂金属原位反应形成富含无机成分的SEI膜,改变了原生钝化层的结构。这些丰富的无机成分通常被认为能够增强SEI的机械强度,使其能够适应反复的体积变化并抑制枝晶生长,从而促进锂金属在低温下的稳定循环。
本文进一步利用AIMD计算来阐明PN添加剂与纯二甲基乙醇胺(DME)中的锂金属之间界面反应的机制(图1f),或混合醚类溶剂体系。图1f展示了不同模拟时间尺度下的AIMD模拟快照。PN被发现自动吸附到锂金属表面,并且NO3–阴离子首先分解,形成Li3N和Li2O组分。与此同时,PAQ+中的S=O键断裂,并在锂金属表面生成Li2S组分。由于反应物通过扩散与更多的Li0接触,PN 通过C-F断裂经历了快速的脱氟过程,导致大量LiF形成。然而,醚溶剂相对于锂金属来说相对稳定,在模拟的时间尺度内,锂金属表面没有发生分解反应。
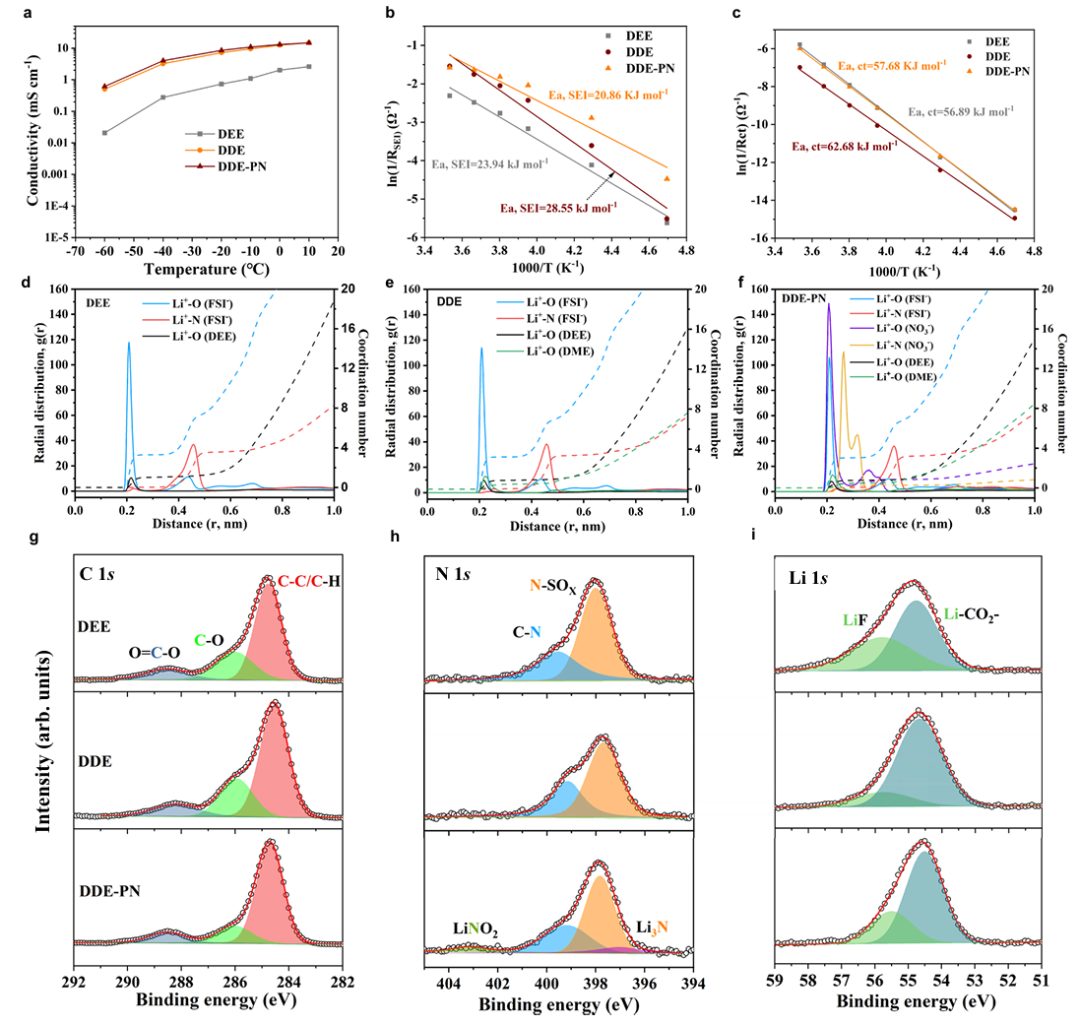
图2 离子扩散、电荷转移、电解质的溶剂化结构以及 SEI的化学成分
此外,还对1.0 M LiFSI/DEE(注释为DEE)、1.0 M LiFSI/DEE+DME(DEE: DME=9:1 体积比,注释为DDE)以及DDE+0.1 M PN(DDE-PN)电解质的离子传输特性、去溶剂化能力以及成膜特性进行了研究和比较。如图 2a所示,使用单一DEE溶剂测量的电解质的离子电导率相对较低,在-40℃时仅为 0.22 mS cm-1,在-60℃时仅为 0.02 mS cm-1,这是由于弱溶剂对锂盐的解离不足所致。
进一步进行了MD模拟,利用径向分布函数来描述平均局部溶质-溶质相互作用环境(图 2d-f)。分析表明,DEE电解质呈现出一种特征性的接触离子对结构,其中Li+的溶剂化壳包含富含FSI–的阴离子,其中几乎没有DEE分子,平均配位数为 3.2 FSI–和 1.0 DEE,这与之前的研究结果一致。引入DME溶剂后,由于Li+与DME之间存在强烈的相互作用,DME进入Li+溶解壳层,取代了一些 DEE 溶剂分子。平均配位数变为 3.2 FSI–、0.88 DEE 和 0.48 DME,这表明DME进入了溶解壳层并挤出了部分DEE溶剂。然而,当PN添加剂被引入混合溶剂体系时,由于NO3–与Li+的结合能力更强,它进入Li+溶解壳层,取代了溶剂。平均配位数变为 3.0 FSI–、0.46 NO3–、0.58 DEE和 0.4 DME。在Li+溶剂化壳层中出现的溶剂总量减少有效地减轻了 DME 对去溶剂化动力学的不利影响。
通过测量不同温度下Li||Li对称电池的电荷转移电阻,确定了不同电解质中Li+的脱溶剂化活化能。图 2c显示,DEE系统和 DDE二元体系的活化能分别为 56.89 kJ mol-1 和 62.68 kJ mol-1。DDE系统中较高的脱溶剂化能是由于DME与Li+的络合作用更强,显著增加了Li+脱溶剂化的难度。然而,PN添加剂引入了NO3–离子,这些离子能够进入第一层溶剂化壳层,从而改变了Li+的溶剂化结构,并削弱了Li+与溶剂之间的相互作用(57.68 kJ mol-1),这与分子动力学模拟结果相符。
为了研究 SEI 形成机制,采用了XPS来表征循环锂金属表面的 SEI 的成分和结构。在DEE中形成的SEI中C-O和C=O的峰值强度明显低于DDE(图 2g),这表明在DEE中溶剂分解受到抑制,而与DDE相比,在DEE中抑制了溶剂分解。因此,在DEE中,SEI主要由阴离子还原产生的大量无机物质组成,而引入DME则破坏了弱溶剂中富含阴离子的溶剂化结构,极大地增加了有机成分的比例(图2h、i)。有趣的是,在DDE-PN电解液中形成的SEI包含更多的无机成分,比如LiF和Li3N,这是因为源自锂金属表面PN分解的富含无机成分的SEI能够有效抑制混合二元溶剂体系中的溶剂分解。
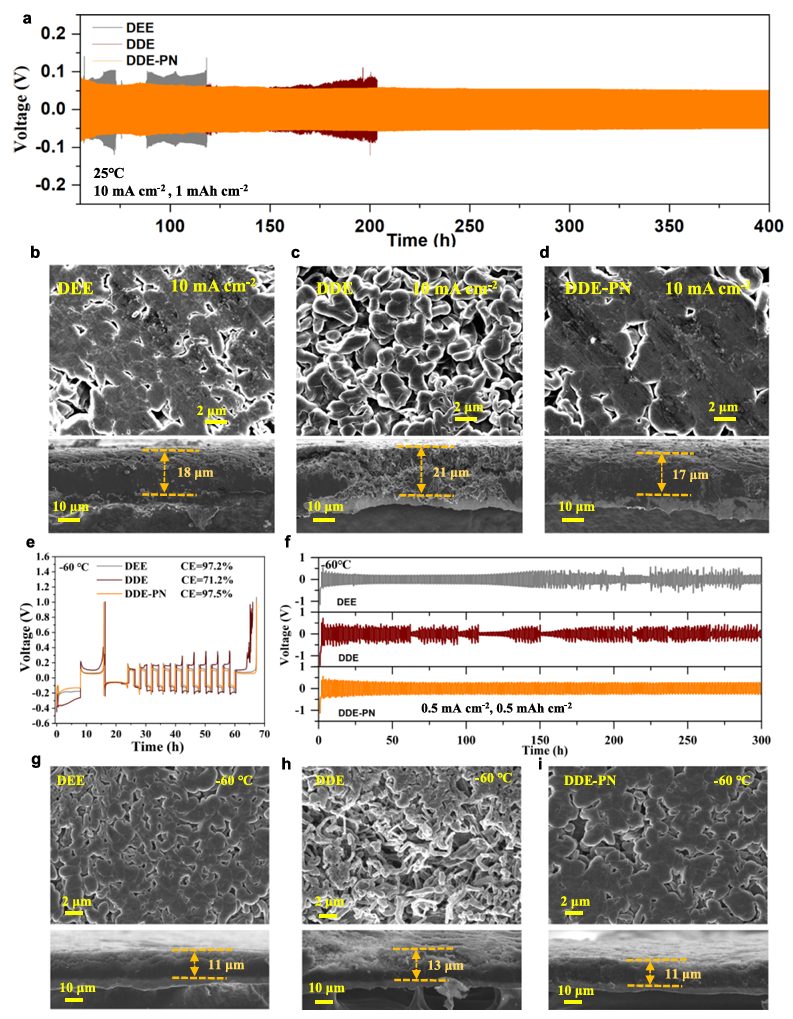
图3 在快速充电和低温条件下锂金属的电化学性能
为了测试锂金属在不同电解质中的长期循环稳定性,组装了Li||Li对称电池。结果发现DDE-PN电解质有效地延长了Li||Li对称电池的寿命(图3a)。特别是在高电流密度下DDE-PN电解质表现出最低的过电位,表明锂金属具有更好的可逆性。不同电流密度下的锂金属沉积形态(图 3b-d)显示,在DEE系统中沉积的锂呈现出均匀分布的块状锂,且几乎没有空隙,在低电流下表现良好。
更重要的是,随着电流密度的增加,DEE和DDE电解质体系中的锂金属倾向于以松散的小颗粒形式沉积,这些颗粒之间存在显著较大的间隙(图3b、c)。即使在电流密度高达10 mA cm-2的情况下,DDE-PN电解质中的锂金属仍形成了紧密(图 3d)且致密的团聚体,这与DDE-PN系统良好的锂离子传输动力学密切相关。
随后,研究了锂金属在低温下的电化学性能。如图3e所示,测量得出的DEE的库仑效率(CE)值在 25℃时为98.5%,在-60°C时为97.2%。对于DDE二元体系,尽管如前所述,在-60℃时电解质的离子电导率较高,但测得的 CE仅为-60℃时的71.2%,这应归因于引入强溶剂二甲基乙醇胺(DME)后导致的去溶剂化能垒增加和不稳定的固体电解质界面(SEI)不稳定。如图3f所示,在-60℃条件下,Li||Li对称电池在使用 DEE电解液进行Li电镀/剥离循环约150小时后发生短路。而使用DDE的Li||Li电池在电镀/剥离初期即发生短路。相比之下,使用DDE-PN的Li||Li电池在电镀/剥离过程中稳定循环了超过 300小时。这种改进归因于离子传导性的增强、降低了解溶能垒以及构建了高质量的SEI,这显著提高了在极低温度下锂金属的长期循环性能。
在-60℃的低温条件下,且容量固定为 1 mAh cm-2时,DEE电解液中的锂金属保持了均匀沉积,未形成枝晶(图3g)。然而,在DDE电解液中,锂金属表面出现了大量分布不均的苔藓状和针状枝晶,这与之前的研究结果一致。横截面SEM图像进一步显示,与DEE电解液相比,DDE中的锂沉积主要表现为从基底底部向顶部表面延伸的枝晶生长(图3h)。

图4 电解质氧化稳定性评估及NCM811结构表征
阻碍醚基稀溶液电解质应用的一个主要障碍在于其在高压下氧化稳定性差的问题,这会降低醚基电解质与高压NMC811正极的相容性。如图 4a 所示,使用铝箔工作电极进行的LSV测量揭示了所测试电解质的不同电化学稳定性边界。传统的基于乙二醇醚(DEE)和二乙二醇醚(DDE)的电解质表现出有限的耐氧化性,其电化学窗口在 4.0 V以下受到限制。与此形成鲜明对比的是,所配制的DDE-PN电解质实现了显著的电压耐受性扩展,在相同的测试条件下将阳极稳定性极限推高至4.5 V。这一稳定效果还通过使用碳包覆铝电极的平行LSV测量得到了进一步证实,在这些测量中,PN改性系统始终保持着增强的电压耐久性。随后,评估了这三种电解质与NCM811正极正极的相容性。首先,测量了静态漏电流。如图4b所示,将正极保持在4.3 V的电位下 10000 秒后DEE和DDE电解质中的漏电流逐渐降低,并分别稳定在约0.02 mA和0.008 mA。相比之下DDE-PN电解质中的漏电流迅速衰减并稳定在仅0.001 mA,这表明其界面电化学稳定性更好。
其次,测试了Li||NMC811电池在三种不同电解质中的自放电特性。在将电池充电至4.3 V并静置20小时后,电池的自放电行为存在显著差异。如图4c所示,在DDE-PN电解质体系中,静置20小时后电池电压稳定在4.21 V。然而,在DDE电解质中,电池电压在20小时后降至3.8 V,在纯DEE电解质中,电池电压在仅12小时内迅速降至3.5 V以下。同样地,采用DDE-PN电解质的Li||NMC811半电池表现出良好的长期循环性能和相对稳定的库仑效率(CE)。如图4d、e所示,在1.0 C下经过400次循环后,采用DDE-PN电解质的Li||NMC811半电池仍保留了其容量的80%,在5.0 C下经过1000次循环后仍保留了76%的容量,这表明在高压下具有可靠的快速充放电能力。相比之下,采用DEE和DDE电解质的电池在初始循环期间无论是在慢速还是快速充放电速率下都表现出快速衰减,因为醚溶剂无法承受如此高的工作电压。
PN添加剂的引入显著抑制了高电压下醚基电解质的分解,从而显著提高了醚溶剂与高电压NMC811正极的相容性。如图4f所示,SEM图像显示,在DEE和DDE电解液中,经过50次循环后,NMC811正极出现严重的破碎现象。颗粒破碎会导致活性物质脱落或电子接触不良,从而导致电池极化加剧、活性物质含量降低以及可逆性下降。相比之下,在 DDE-PN 电解液中循环后,正极颗粒基本保持完整。图4g中的HRTEM图像也显示,在设计的电解液中循环后的NMC811正极表面的阴极电解质界面(CEI)更薄、更致密,并且二次粒子的完整性更好,没有明显的裂纹。
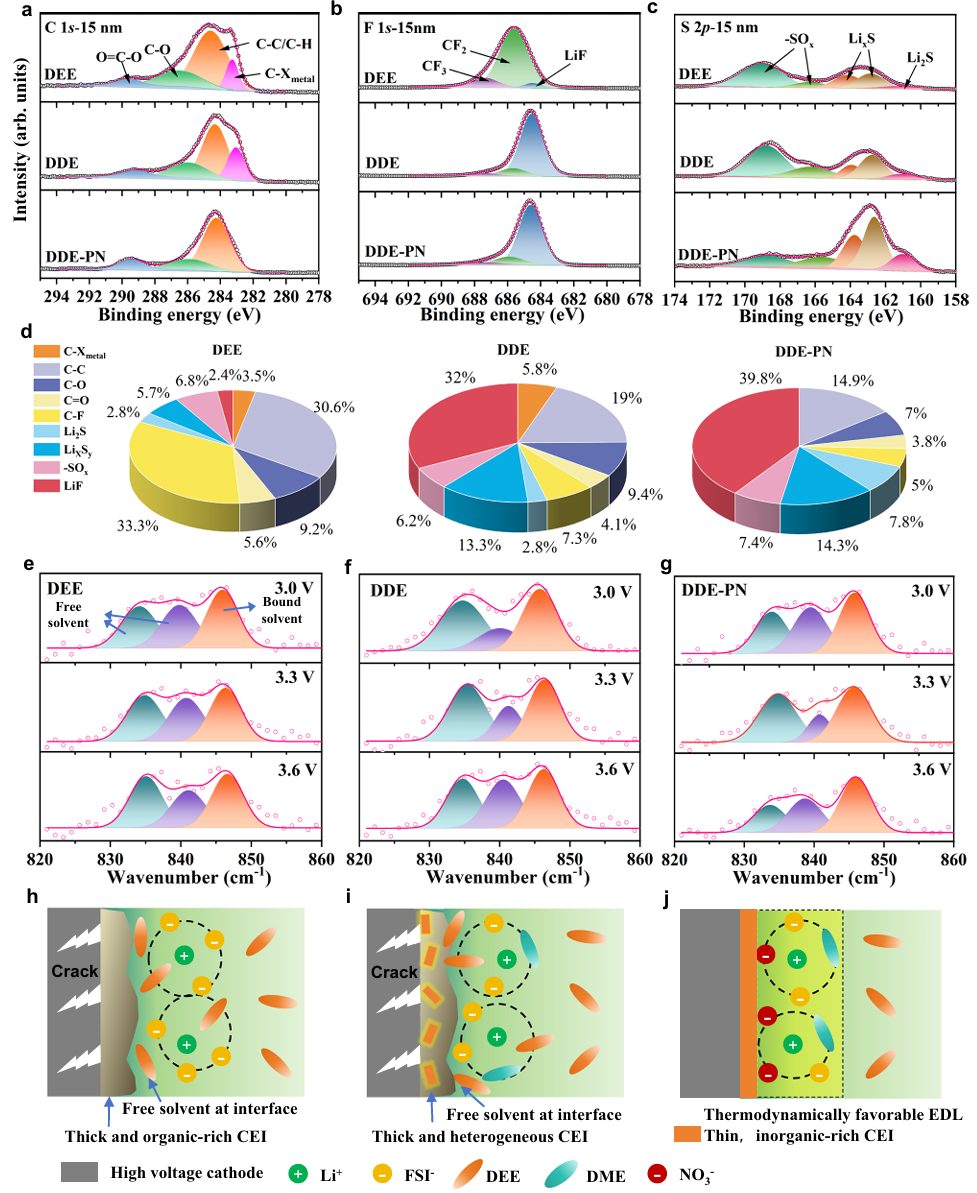
图5 正极表面CEI组成及电解质结构的表征
为了探究醚基电解质与高电压NMC811电池的兼容性得以显著提升的原因,进行了深入的XPS分析,以检测在不同电解质中经过 50 次循环后NMC811材料表面形成的CEI的化学成分。在DEE和DDE中,在C 1s光谱中观察到了一个位于283.1 eV的新峰,对应于C-X金属键,这表明在NMC811界面处发生了严重的电解质分解。随着溅射过程的推进,在 DDE-PN中观察到 C 1s 强度显著降低,同时在 DDE-PN 电解液形成的 CEI 的内层中发现了更多的无机化合物,如 LiF、Li2S 和 LixSy。值得注意的是,由 DDE-PN 电解液生成的 CEI 出现了最强的 LiF 峰,其内部 LiF 含量超过 39.8%,而由 DDE 和 DEE 电解液形成的 CEI 的内部 LiF 含量分别为 32%和 2.4%(图 5d)。CEI内从有机组分到无机组分的梯度分布赋予了有效的电子阻挡能力,减少了电子对CEI的渗透,从而确保了电池的循环稳定性。
在800~900 cm-1范围内存在多个拉曼峰。821和849 cm-1处的谱峰对应不同异构体的自由醚溶剂,879 cm-1处的谱峰对应Li+溶剂化的醚溶剂。自由溶剂峰与配位溶剂峰的面积比反映了溶剂的边界状态占总体积的比例(图5e-g)。通过拟合配位醚溶剂的比例与正极/电解质界面电压的关系,在不添加PN添加剂的电解质中,正极界面上的大多数醚分子以自由状态存在,Li+溶剂化的醚溶剂比例随着电压的升高而降低。
因此,由大量负极不稳定的游离醚溶剂组成的EDL在高压下无疑会降解。而在DDE-PN电解质体系中,随着电压的升高,Li+溶剂化醚溶剂的比例显著增加,说明NO3–的引入使得更多的醚参与到溶剂化壳中,形成了能够在正极承受高压环境的阳极稳定EDL。因此,NO3–的优先吸附导致溶剂的位移,并且由于NO3–和Li+之间的强相互作用,Li+被大量吸引到电极界面上。因此,EDL层中存在的醚与Li+广泛结合,构建了高度氧化稳定的EDL。因此,通过降低初始形成的EDL中活性醚溶剂的含量,PN添加剂能够改变正极界面上醚溶剂的稳定性。总的来说,DDE-PN电解质通过形成薄的、富含无机的CEI和建立热力学有利的EDL提供了双重保护机制。
这有效减轻了醚基电解质分解和正极材料结构降解,显著增强了醚基电解质与高压正极的相容性。因此,通过降低初始形成的EDL中活性醚溶剂的含量,可以将高压正极降解游离溶剂的风险降至最低。此外,这种无溶剂、富阴离子的EDL促进了富无机CEI的形成,通过双重保护,显著提高了醚基电解质体系中正极/电解质界面的电化学稳定性(图5h-j)。

图6 低温下全电池和软包电池的电化学性能
最后,分别在Li||NMC811全电池和实际LMB中研究了这三种电解质的电化学性能。循环在室温和超低温条件下进行。如图6a所示,DDE-PN电解质使Li(40µm)||NMC811(3.0 mAh cm-2)全电池在室温下表现出良好的循环性能,300次循环后容量保持率为84%。相比之下,DDE和DEE电解质表现出较差的循环性能,由于醚基溶剂的高压兼容性不理想,在初始循环和循环70次后分别出现严重的容量下降。此外,在恶劣的超低温条件下(-60℃), Li||NMC811全电池能够稳定地充放电,在100次循环中提供110 mAh g-1的高可逆容量,容量保持率为93.3%,表明稳定的循环稳定性(图6c)。相应的随温度变化的充放电曲线也呈现出明显的电压平台,表明低温下具有良好的充放电能力(图6d)。
此外,500 mAh Li||NMC811软包电池也在DDE-PN电解质中进行了超低温测试。如图6e所示,在-60℃、-80℃和-85℃下,软包电池的能量密度分别为337.3 mAh、257.4 mAh和245.4 mAh,相对于室温,容量保持率分别为66.1%、50.5%和48.1%。令人兴奋的是,即使在-85℃下,软包电池也可以达到171.8 Wh kg-1的比能量。使用DDE-PN电解质的软包电池在-50℃下可以以3.0 C的高倍率放电,在如此恶劣的低温条件下显示出938.5 W kg-1的比功率(图6f)。
Multifunctional electrolyte additive for high power lithium metal batteries at ultra-low temperatures,Nature Communications,2025.
https://www.nature.com/articles/s41467-025-58627-3

电解液是决定锂电池性能的关键成分,特别是在快速循环和低温等极端条件下。然而,传统的电解质设计原理通常依赖于溶剂、盐和功能添加剂的均匀混合物,无法同时满足锂金属电池对负极/正极界面稳定性和离子传输动力学的要求。
清华大学刘凯等人提出了一种自分隔电解质设计方法。LiTDI具有在正极/电解质界面上选择性自组装的能力,将电解质分隔成异质电解质。靠近阴极侧,LiTDI可以诱导出一个界面高浓度区,在该区域中,富含阴离子的溶剂化结构有利于形成稳定的正极-电解质界面相(CEI)。在整体上,电解质保持低浓度和低粘度,确保快速离子传输和优越的速率性能。Li||NCM811电池实现超过500次稳定循环,容量保持率为80.3%,在10C放电速率下提供169.3 mAh g-1。在低温条件下(-20℃),电池在0.5C充放电条件下保持了出色的稳定性,超过700次循环,容量保持率为96.6%,平均库仑效率为99.2%。这项工作提供了一种新的电解质设计范式,解决了LMBs在高压和低温应用中的关键挑战。
相关工作以《Self-Compartmented Electrolyte Design for Stable Cycling of Lithium Metal Batteries Under Extreme Conditions》为题在《Angewandte Chemie International Edition》上发表论文。
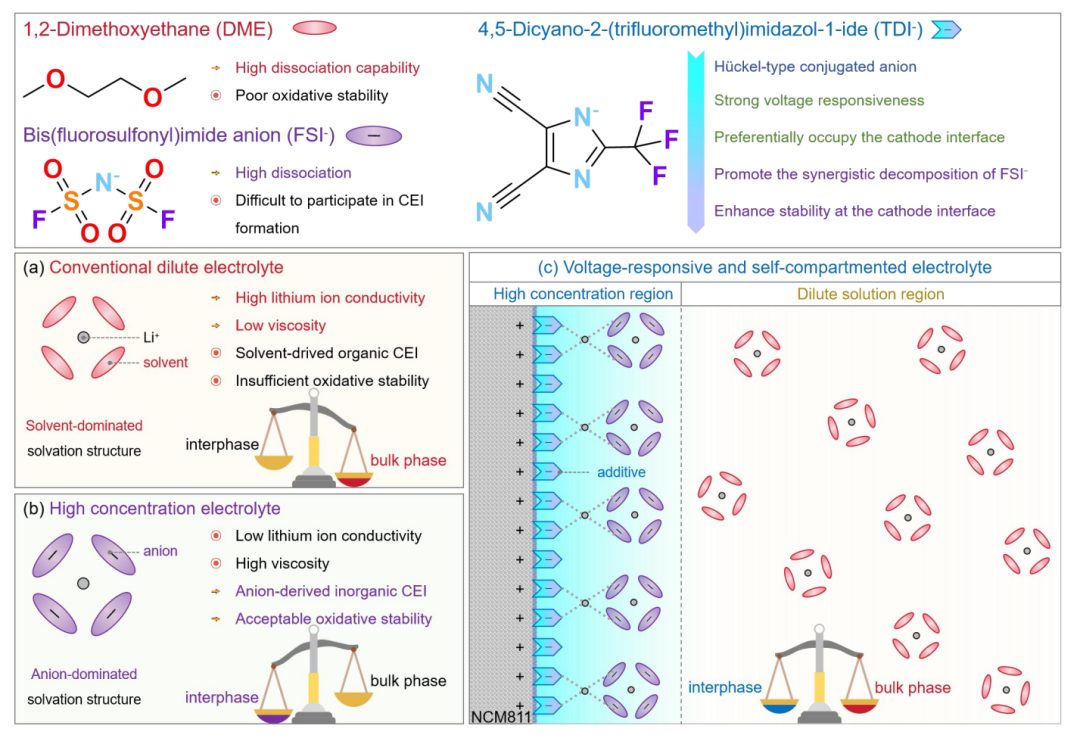
图1 LiTDI增强电压响应间隔电解质系统概述
在这项工作中,引入了一种新的“自分隔电解质”策略,以解决传统电解质设计中遇到的高稳定/低电阻界面与低电阻体电解质之间的矛盾。采用与正极表面配位能力强的不对称TDI–阴离子作为基础电解质的添加剂。这种设计背后的整体工作机制如图1所示,图1比较了不同电解质配置下的溶剂化结构。在低浓度电解质中(图1a),溶剂分子主导了Li+的溶剂化结构,确保了大块溶液的高离子电导率,同时损害了CEI的稳定性。另一方面,在高浓度电解质(图1b)中,阴离子在溶剂化结构中占主导地位,增强了CEI稳定性,但以牺牲体积电导率为代价,导致动力学势垒增加。
相比之下,LiTDI增强电解质,即在基础电解质中添加0.2 M LiTDI(表观浓度)(图1c),有效地结合了这两种体系的优点。LiTDI专门积聚在正极表面,有利于形成高浓度、无溶剂、富含阴离子的界面溶剂化结构。然而,散装电解质保持低浓度状态,具有低粘度和高离子电导率。
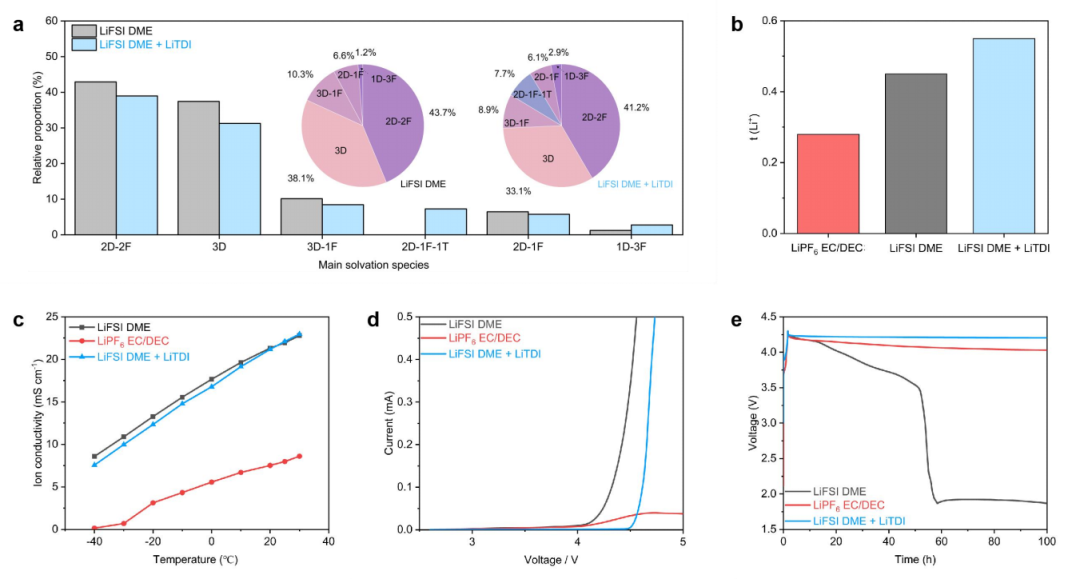
图2 LiTDI对醚基电解质体溶剂化结构和电化学性能的影响

图3 LiTDI对Li||NCM811电池电化学性能的影响
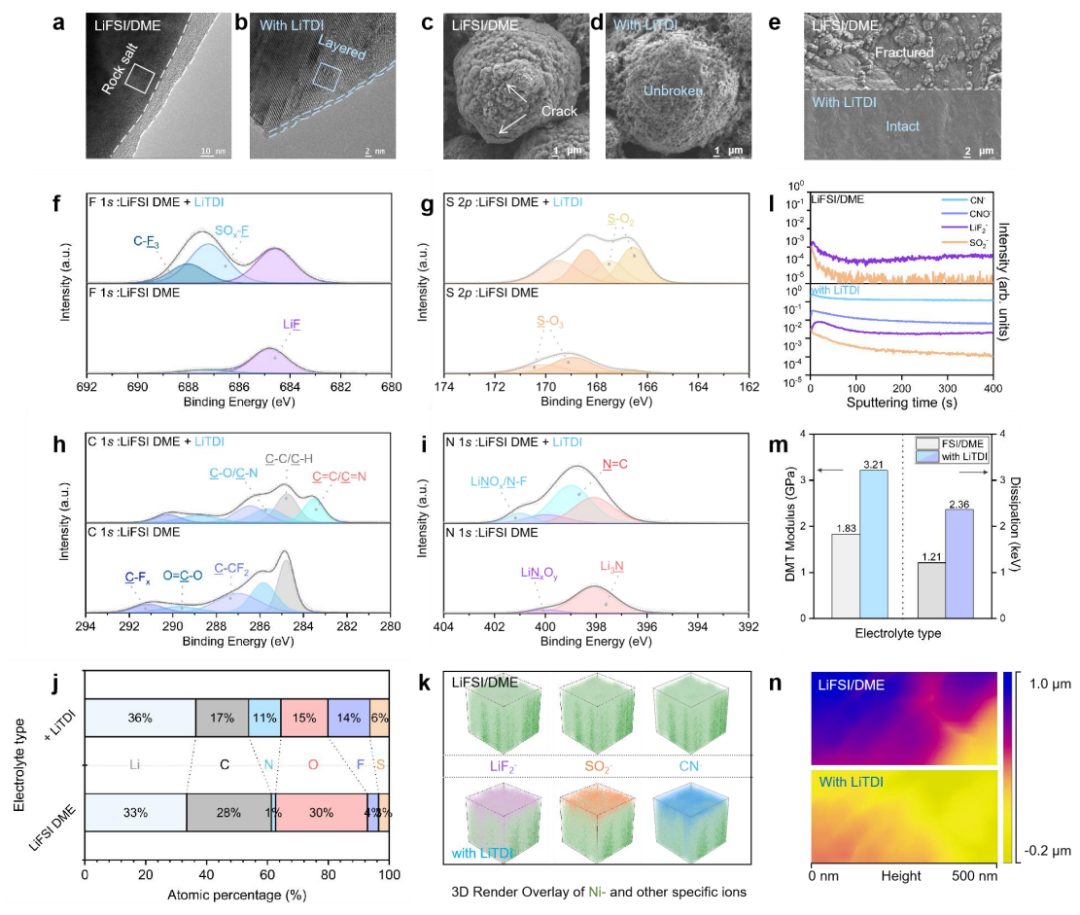
图4 正极和CEI的结构、组成和力学特性
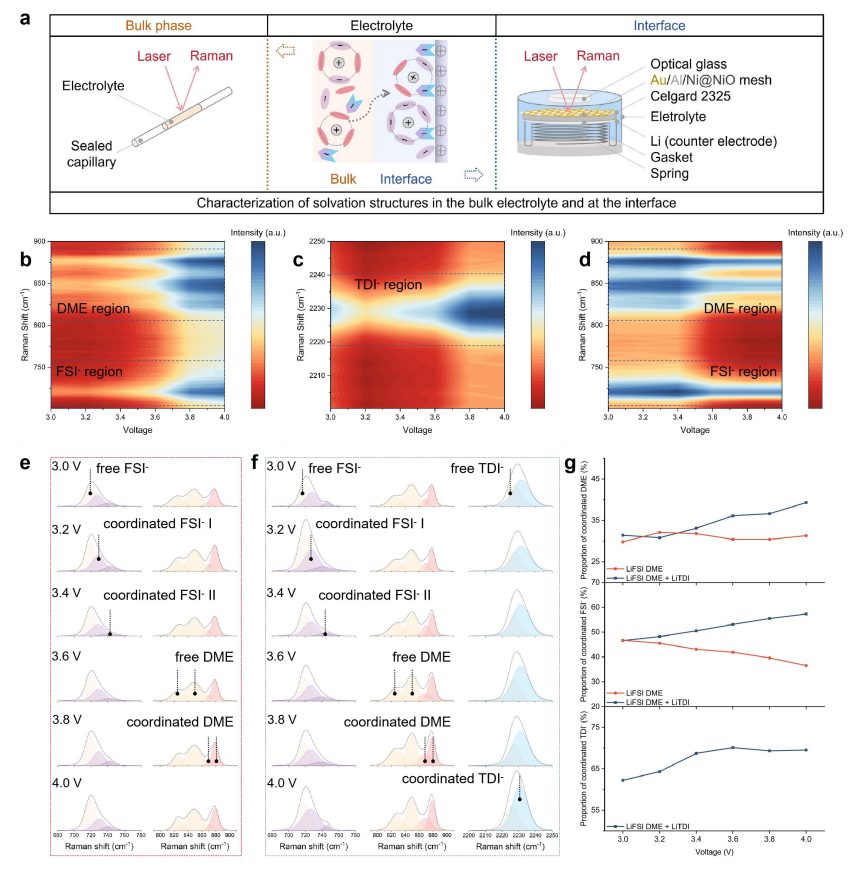
图5 不同条件下LiTDI增强电解质的界面和体相溶剂化结构
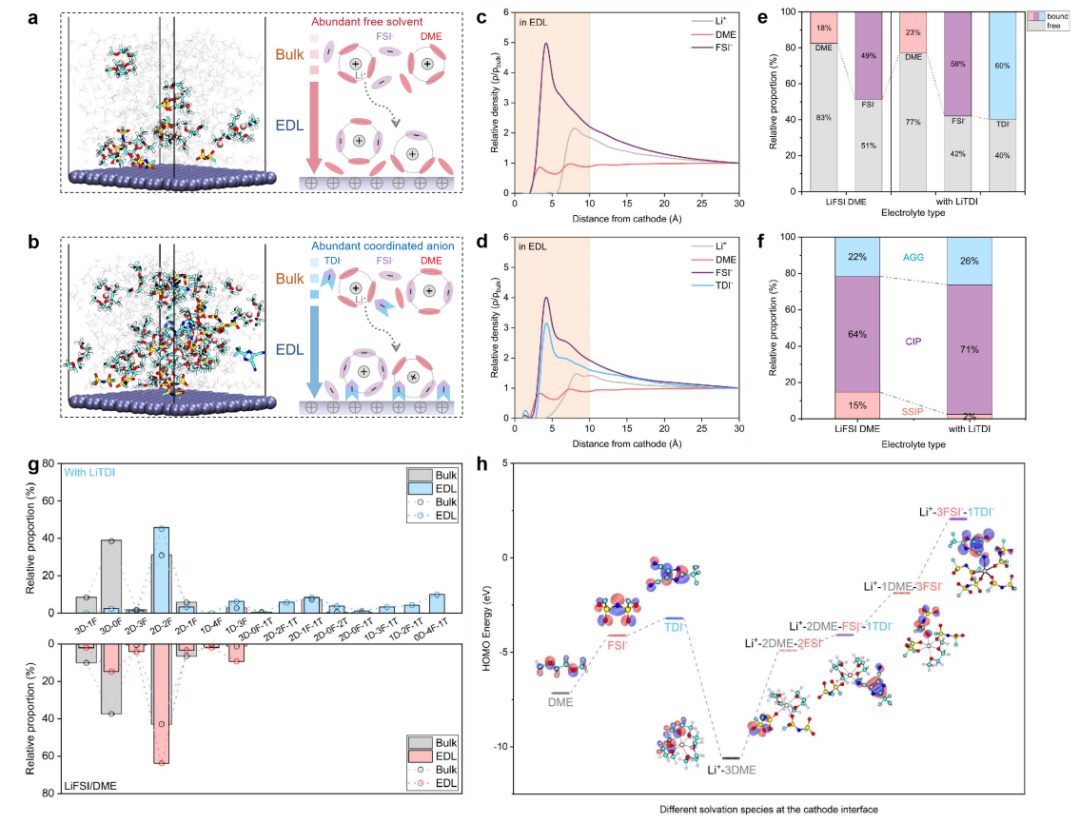
图6 分子动力学模拟和统计溶剂化结构分析
利用分子动力学(MD)模拟分析了电化学平衡条件下的双电层(EDL),为正极侧附近溶剂化物质的重排提供了新的见解。碱性电解质呈现出阴离子和阳离子的分散分布。相比之下,LiTDI增强的电解质形成了一个更致密的界面层,富含TDI–。对Li+ 3 Å内离子种类的统计分析(图6a-b)进一步阐明了阴极界面的特定溶剂化结构。在碱电解质中(图6a),游离二甲醚分子和FSI–主导EDL,形成溶剂分离的溶剂化结构。然而,当LiTDI存在时(图6b),溶剂化环境发生变化,TDI–成为界面处的主导阴离子。
TDI–与Li+的强配位不仅限制了游离二甲醚的积累,而且促进了更紧密离子结构的形成。径向密度分布图(图6c-d)证实了这些趋势,显示在LiTDI系统中,FSI的概率密度降低,为4.23 Å,同时TDI穿透更靠近电极的可能性更高(1.36 Å)。TDI–的增加与Li+浓度的增加相关,因为TDI–的积累增强了与锂离子的局部配位。界面附近升高的Li+密度反过来又吸引了额外的FSI离子,导致FSI–出现明显的概率峰值,约为1.51 Å。
吸附能计算进一步支持了这些观察结果,表明与DME相比,TDI–和FSI–与NCM811表面的相互作用更强,这与它们在正极界面的优先积累一致。基于这些空间趋势,图6e量化了两种电解质中DME、FSI–和TDI–的自由态和配位态的相对比例。在基础电解质中,18%的二甲醚和49%的FSI–处于配位态,而大多数保持自由态。相比之下,LiTDI增强电解质的DME和FSI–的配位分数均显著增加,其中DME配位分数上升至23% 622,FSI–配位分数上升至58%。
Self-Compartmented Electrolyte Design for Stable Cycling of Lithium Metal Batteries Under Extreme Conditions,Angewandte Chemie International Edition ,2025.
https://onlinelibrary.wiley.com/doi/10.1002/anie.202504632