

在材料科学研究的微观世界里,深入理解原子间电荷的分布与转移情况,对于揭示材料的诸多性质和化学反应机制至关重要。今天,就为大家带来一篇干货满满的教程,详细介绍如何使用 VASP 计算 Bader 电荷。
Bader电荷分析:原理与科学意义
Bader电荷分析是一种基于电子密度拓扑结构的原子电荷划分方法,由理论化学家Richard Bader于20世纪90年代提出,现已成为材料模拟领域的核心分析工具之一。其核心思想是通过电子密度梯度场的临界点(Critical Points)划分原子区域,将总电子密度分配到各个原子上,从而精确描述电荷分布与转移行为。这一方法不仅突破了传统电荷划分的局限性,还为理解化学键本质、界面相互作用及反应机理提供了全新的视角。
1. 物理意义明确:电子密度拓扑特征的化学直观性
Bader电荷的核心优势在于其物理意义的明确性。传统电荷划分方法(如Mulliken电荷)高度依赖基组的选择,计算结果可能因基组类型或计算参数的不同而产生显著偏差。而Bader电荷基于电子密度的拓扑特征,通过数学上严格定义的零通量面(Zero-Flux Surface)划分原子区域,完全独立于基组或赝势的选择。例如,在金属–有机框架材料中,Bader电荷能清晰显示金属节点与有机配体间的电荷转移方向与幅度,而Mulliken电荷可能因基组重叠问题导致结果失真。这种独立性使得Bader电荷更贴近化学直觉,成为解释实验现象(如XPS化学位移)的有力工具。
2. 电荷转移量化:从微观电子结构到宏观性能的桥梁
Bader电荷的量化能力使其在复杂体系中展现出独特价值。以异质结界面或催化吸附体系为例,Bader电荷可以精确计算界面处的电荷再分布,揭示电子转移的驱动力与路径。例如:
· 催化反应:在CO₂还原反应中,Bader电荷分析显示催化剂表面的活性位点(如Cu单原子)通过电子转移活化CO₂分子,形成关键中间体*COOH,其电荷变化与反应能垒直接相关。
· 电池材料:在锂离子电池正极材料(如LiCoO₂)中,Bader电荷可量化锂脱嵌过程中Co的氧化态变化,关联电荷转移与容量衰减机制。
这些应用表明,Bader电荷不仅是理论计算的输出结果,更是连接电子结构理论与宏观材料性能的关键纽带。
3. 广泛应用场景:多尺度材料体系的普适性工具
Bader电荷的拓扑划分方法具有极强的普适性,适用于从分子到晶体、从表面到界面的各类体系:
· 分子体系:分析小分子(如H₂O、NH₃)的极性键电荷分布,解释偶极矩与反应活性。
· 表面与界面:研究金属氧化物表面吸附分子(如O₂、H₂O)的电荷转移,揭示表面重构或催化活性位点的形成机制。
· 低维材料:在石墨烯、过渡金属硫化物(如MoS₂)中,Bader电荷可量化边缘态或缺陷态的电荷局域化效应,指导能带工程设计。
这种广泛的适用性使其在催化、能源存储、半导体器件等领域成为不可或缺的分析手段。
VASP与Bader电荷的协同优势
VASP(Vienna Ab-initio Simulation Package)作为第一性原理计算的标杆软件,凭借其高精度的电子密度计算能力,为Bader电荷分析提供了坚实的数值基础。其核心优势体现在以下方面:
1. 高精度电荷密度输出
VASP通过求解Kohn-Sham方程,能够输出高分辨率的电荷密度文件(CHGCAR),确保Bader电荷分析的准确性。例如,在复杂表面吸附体系中,VASP的赝势(如PAW方法)与高精度基组(平面波展开)可精确描述电荷密度的空间振荡,避免因数值误差导致的原子区域划分偏差。
2. 灵活的参数兼容性
VASP支持用户自定义电荷密度网格(通过NGXF、NGYF、NGZF参数),允许根据体系尺寸与计算资源灵活调整精度。对于大体系(如纳米颗粒或超胞模型),适当降低网格密度可在保证结果合理性的同时显著节省计算成本。
3. 与后处理工具的无缝对接
VASP生成的电荷密度文件(CHGCAR、AECCAR0、AECCAR1)可直接用于Bader分析程序,无需复杂格式转换。通过简单的脚本操作(如chgsum.pl合并文件),用户可快速完成电荷密度预处理,显著提升分析效率。
接下来,就将手把手详细教你如何在 VASP 中进行 Bader 电荷计算的具体操作,让你能够轻松掌握这一强大的分析工具,开启材料微观世界深入探索的大门。

一、准备工作
1. 软件安装
o VASP安装与配置
VASP(Vienna Ab-initio Simulation Package)是进行第一性原理计算的核心工具,其安装需遵循以下步骤:
1. 获取许可证:VASP为商业软件,需通过官方渠道获取许可证并下载源代码(通常包括vasp_std、vasp_gam等可执行文件)。
2. 编译安装:
o 解压源代码后,根据计算平台(如Intel或GNU编译器)修改makefile.include配置文件。
o 安装依赖库(如FFTW、MPI),运行make all编译生成可执行文件。
3. 环境变量配置:
o 将VASP可执行文件路径(如/opt/vasp/bin)添加到$PATH环境变量:
exportPATH=$PATH:/opt/vasp/bin
o 验证安装:在终端输入vasp_std,若无报错提示“Error opening input files”,则安装成功。
o Bader电荷分析程序安装
Bader电荷分析程序需独立安装以处理VASP输出的电荷密度文件:
1. 下载与解压:
o 访问官方页面(https://theory.cm.utexas.edu/henkelman/code/bader/)下载最新版本(如bader.tar.gz)。
o 解压文件:
tar-zxvf bader.tar.gz
2. 编译与配置:
o 进入解压目录,运行make编译生成可执行文件bader。
o chgsum.pl脚本获取
chgsum.pl是合并电荷密度文件的关键脚本,需从VTST工具包官网获取:
1. 下载地址:访问VTST官网(https://theory.cm.utexas.edu/vtsttools/),下载vtstscripts.tar.gz并解压。
2. 脚本存放:将chgsum.pl复制至工作目录,或添加其路径至$PATH以便全局调用。
2. 计算文件准备
o POSCAR:结构文件
POSCAR需精确描述计算体系的原子坐标与晶胞信息:
1. 格式要求:
o 第一行为注释(如体系名称)。
o 第二行为晶胞缩放因子(通常为1.0)。
o 第三至五行定义晶胞基矢(单位:Å)。
o 第六行列出元素种类,第七行为各元素原子数。
o 后续行设置原子坐标(笛卡尔坐标或分数坐标,由Selective dynamics行指定)。
2. 校验:检查POSCAR合理性,避免原子重叠或晶胞畸变。
o INCAR:参数控制文件
INCAR需包含以下关键参数以确保Bader电荷分析的准确性:
1. 电荷密度输出设置:
LAECHG = .TRUE. # 输出AECCAR0(核心电子密度)和AECCAR1(价电子密度)
LCHARG = .TRUE. # 输出总电荷密度文件CHGCAR
2. FFT网格密度优化:
NGXF = 200 # X方向网格点数(根据晶胞尺寸调整,建议≥200)
NGYF = 200 # Y方向网格点数
NGZF = 200 # Z方向网格点数
o 注意事项:网格密度不足可能导致电荷密度插值误差,影响Bader划分精度;过大则显著增加计算耗时。
o KPOINTS:k点网格设置
1. 网格类型选择:
o 对于周期性体系,推荐Monkhorst-Pack网格;分子体系可选用Gamma中心网格。
2. 密度建议:
o 块体材料:6×6×6或更高。
o 表面或分子:根据真空层厚度调整,如3×3×1。
o POTCAR:赝势文件
1. 文件生成:
2. 版本一致性:确保所有赝势文件来自同一泛函(如PBE或LDA),避免混合使用导致计算误差。
二、VASP计算
1. 输入文件准备与目录管理
在进行VASP计算前,需确保所有输入文件正确配置并存放于同一工作目录,以避免路径错误或文件缺失问题:
· 文件清单与作用:
o POSCAR:定义计算体系的晶胞参数、原子坐标及元素种类。
o INCAR:设置计算参数(如电子步收敛标准、电荷密度输出选项)。
o KPOINTS:指定k点网格类型与密度,影响布里渊区积分精度。
o POTCAR:包含所有元素的赝势文件,决定电子–离子相互作用的描述精度。
· 目录结构建议:
o 创建独立文件夹(如Bader_Calculation),将所有输入文件置于其中。
o 避免与其他计算任务混用目录,防止文件覆盖或混淆。
2. 运行VASP计算
根据计算平台的不同,VASP可通过作业调度系统或本地命令行启动:
o 使用PBS作业提交系统
PBS(Portable Batch System)是超算中心常用的任务调度工具,其典型操作流程如下:
1. 编写作业脚本:
创建名为vasp.sh的脚本文件,内容示例:
#!/bin/bash
#PBS -N Bader_Job # 作业名称
#PBS -l nodes=2:ppn=24 # 申请2个节点,每节点24核
#PBS -l walltime=24:00:00 # 最大运行时间24小时
cd$PBS_O_WORKDIR # 进入工作目录
mpirun -np48 vasp_std # 并行运行vasp_std,使用48核
2. 提交任务:
qsub vasp.sh
o 查看任务状态:qstat -u [用户名]。
o 终止任务:qdel [作业ID]。
o 本地直接运行
在本地服务器或工作站上,可通过MPI并行命令直接启动VASP:
mpirun -np16 vasp_std # 使用16核并行计算
· 参数说明:
o -np 16:指定使用的CPU核心数,需根据硬件资源合理分配(通常为物理核心数的70-80%)。
o vasp_std:标准版本的VASP可执行文件,适用于大多数计算任务。
· 注意事项:
o 确保已正确配置MPI环境(如OpenMPI或Intel MPI)。
o 对于大体系(>100原子),建议使用vasp_gam(Gamma点优化版本)以节省计算时间。
3. 计算完成与输出文件解析
成功运行后,工作目录中将生成以下关键文件:
o AECCAR0
· 内容:记录体系中所有原子的核心电子(Core Electron)密度分布。
· 作用:在Bader分析中,需将核心电子密度与价电子密度合并,以还原总电子密度。
· 文件格式:与CHGCAR格式一致,包含三维网格点的电子密度值。
o AECCAR2
· 内容:记录价电子(Valence Electron)密度分布,反映化学键与电荷转移行为。
· 合并操作:使用chgsum.pl脚本将AECCAR0与AECCAR2相加,生成总电荷密度文件CHGCAR_sum:
chgsum.pl AECCAR0 AECCAR2
o CHGCAR
· 内容:包含体系的总电子密度(核心电子 + 价电子),是Bader电荷分析的直接输入。
· 校验要点:
o 检查文件末尾是否包含完整的密度数据(避免因计算中断导致文件截断)。
4. 计算状态验证与错误排查
为确保计算成功,需检查以下关键输出文件:
· OUTCAR:
o 搜索General timing and accounting,确认计算正常结束。
o 检查reached required accuracy标志,确保电子步收敛。
· OSZICAR:
o 查看离子步迭代次数与能量变化,确认结构优化或静态计算收敛。
· 常见错误处理:
o 电荷密度不收敛:在INCAR中降低EDIFF(如EDIFF=1E-5)或增加NELM(最大电子步数)。
o 内存不足:减少并行核数或增加节点内存分配。

三、Bader电荷分析
1. 准备Bader分析环境
在进行Bader电荷分析前,需确保Bader程序的可执行文件与VASP输出文件位于同一目录,以避免路径错误或权限问题:
1. 程序部署:
o 将编译好的bader可执行文件(通常位于Bader源码目录的bader子文件夹)复制至包含CHGCAR、AECCAR0、AECCAR2的工作目录。
o 验证权限:通过chmod +x bader赋予可执行权限,确保可直接调用。
2. 脚本依赖检查:
o 确认chgsum.pl脚本已存放于当前目录或系统路径中,可通过which chgsum.pl检查其可用性。
2. 合并电荷密度文件
Bader分析需要总电子密度(CHGCAR)与参考电荷密度(CHGCAR_sum)进行比对,具体操作如下:
1. 生成参考文件CHGCAR_sum:
o 运行命令:
chgsum.pl AECCAR0 AECCAR2
o 脚本功能:
2. 校验合并结果:
o 检查CHGCAR_sum文件头中的网格密度信息(与CHGCAR对比),确认两者匹配。
o 若出现错误,需重新运行VASP计算并确保INCAR中NGXF/NGYF/NGZF参数一致。
3. 执行Bader电荷分析
通过以下命令启动Bader电荷划分:
./bader CHGCAR -ref CHGCAR_sum
参数解析:
· CHGCAR:VASP输出的总电荷密度文件,包含核心与价电子密度之和。
· -ref CHGCAR_sum:指定参考电荷密度文件(即AECCAR0 + AECCAR2),用于校正电荷密度分布。
关键输出文件:
1. ACF.dat:
o 内容格式:
# X(A) Y(A) Z(A) CHARGE MIN DIST
1 8.5022 5.6731 2.9483 +1.234 0.023
2 3.2176 7.8912 1.4567 -0.567 0.018
o 数据解读:
2. BCF.dat:
o 内容格式:
# X(A) Y(A) Z(A) VOLUME ATOM
1 8.5022 5.6731 2.9483 12.34 1
2 3.2176 7.8912 1.4567 8.90 2
o 数据解读:
3. AtomVolumes.dat:
o 内容格式:
# ATOM VOLUME
1 12.34
2 8.90
o 作用:简洁列出每个原子的体积信息,便于快速统计或绘图。
4. 常见问题与解决方案
1. 错误提示“Grid sizes do not match”:
o 原因:CHGCAR与CHGCAR_sum的网格参数(NGXF/NGYF/NGZF)不一致。
o 解决:重新运行VASP计算,确保INCAR中网格参数一致,并重新生成AECCAR0、AECCAR2及CHGCAR。
2. ACF.dat中电荷值异常(如全为0):
o 原因:VASP计算未正确输出电荷密度文件(如LCHARG=.FALSE.)。
o 解决:检查INCAR设置,确保LAECHG=.TRUE.和LCHARG=.TRUE.,重新运行计算。
3. 原子区域边界模糊(MIN DIST过大):
o 原因:电荷密度网格过疏(NGXF/NGYF/NGZF设置过小)。
o 解决:增加NGXF/NGYF/NGZF至更高值(如300),重新计算并分析。
5. 结果验证与应用示例
1. 电荷守恒验证:
o 计算所有原子Bader电荷之和,中性体系应接近0(如总和为±0.1 e以内)。
o 示例:若体系总电荷为+1 e(如阳离子),则Bader电荷总和应接近+1 e。
2. 化学键分析:
o 离子键:电荷转移显著(如Na⁺电荷≈+0.9 e,Cl⁻电荷≈-0.9 e)。
o 共价键:电荷分布较均衡(如C-C键中每个C原子电荷≈±0.2 e)。
3. 催化活性关联:
o 在Pt-Co合金催化剂中,Co原子的Bader电荷(如+0.5 e)反映其向Pt的电子转移,削弱CO吸附强度,提升抗中毒能力。
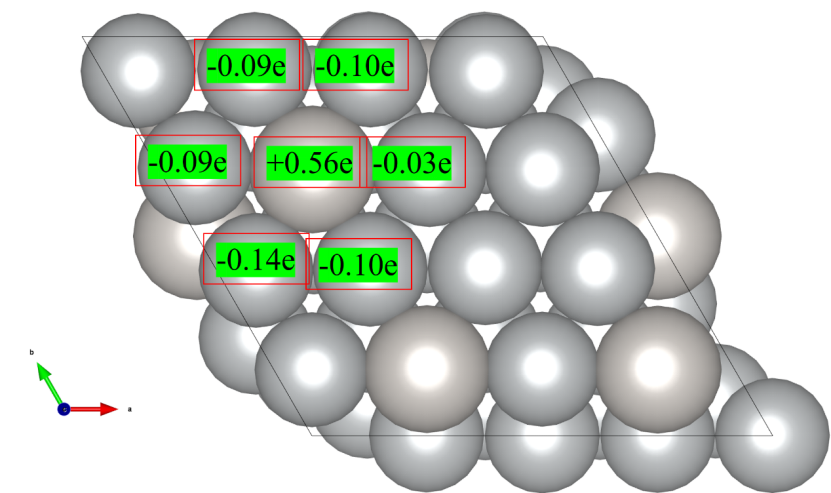
四、结果分析
1. 打开ACF.dat文件,可以看到类似以下的输出:
# X Y Z CHARGE MIN
1 7.0857 4.6600 4.7999 10.0862 1.325
2 7.0857 7.9900 4.7999 9.9138 1.325
3 7.0857 4.6600 8.1301 0.9567 1.325
4 7.0857 7.9900 8.1301 1.0433 1.325
o 第一列为原子序号。
o 第二、三、四列为原子坐标。
o 第五列为原子的Bader电荷。
o 第六列为原子到Bader电荷体积边界的最小距离。
2. 根据你的研究目的,可以对Bader电荷数据进行分析,例如:
o 计算不同原子之间的电荷转移。
o 分析化学键的离子性/共价性。
o 研究吸附分子与表面的电荷转移。
五、注意事项
1. FFT网格密度: FFT网格密度越大,Bader电荷分析结果越精确,但计算量也越大。建议进行收敛性测试,选择合适的FFT网格密度。
2. 赝势:不同的赝势可能会对Bader电荷计算结果产生影响。建议使用高质量的赝势。
3. Bader电荷分析程序的版本:不同版本的Bader电荷分析程序可能会有不同的输出格式。建议使用最新版本。
六、总结
通过本教程的详细解析,我们系统性地介绍了使用VASP进行Bader电荷分析的全流程,从软件安装、参数设置到电荷密度计算与后处理分析,每一步均紧扣实际操作的痛点与关键点。Bader电荷分析作为连接电子密度分布与化学直观理解的核心工具,其价值不仅在于提供定量的电荷转移数据,更在于为材料设计、催化机理研究、界面相互作用分析等提供深层次的物理化学洞察。以下是对本教程核心内容的总结与延伸思考:
1. 流程的严谨性:从输入到输出的闭环验证
Bader电荷分析的可靠性高度依赖于计算流程的严谨性:
· 输入文件校验:确保POSCAR的原子坐标精确、INCAR中NGXF/NGYF/NGZF参数合理、KPOINTS密度足够,是获得高精度电荷密度的前提。
· 计算状态监控:通过检查OUTCAR中的收敛标志(如reached required accuracy)和OSZICAR的能量变化曲线,可有效避免因未收敛导致的电荷密度失真。
· 结果交叉验证:利用ACF.dat的总电荷守恒性(如中性体系电荷和接近0)和文献对比(如H₂O中O原子电荷约-1.2e),可快速判断分析结果的合理性。
2. Bader电荷的科学价值与应用边界
Bader电荷的独特优势使其在多个领域展现不可替代性:
· 催化活性位点识别:通过量化活性位点(如金属单原子或缺陷位)的电荷转移,可关联电子结构变化与催化性能(如过电势、选择性)。
· 界面电荷分离分析:在异质结或吸附体系中,Bader电荷揭示界面处的电子再分布,为光催化、电化学器件设计提供理论依据。
· 化学键本质解析:区分离子键(大电荷转移)与共价键(小电荷转移),辅助理解材料的力学、光学或输运特性。
然而,其局限性亦需注意:
· 静态分析的局限:Bader电荷反映基态电子分布,无法直接描述动态过程(如反应过渡态或激发态)。
· 对网格密度的敏感性:低网格密度(如NGXF=100)可能导致原子区域划分偏差,需通过收敛性测试确定最佳参数。
3. 常见问题的根源与解决策略
· 电荷密度文件不匹配:根源多为VASP计算中NGXF/NGYF/NGZF设置不一致,或合并脚本chgsum.pl执行错误。解决方案是统一参数并重新生成文件。
· 原子电荷异常值:可能源于结构未优化(原子位置偏移)或赝势选择不当(如PAW赝势未包含特定电子态)。需重新优化结构或更换赝势。
· 计算资源与精度的平衡:对于大体系(如纳米颗粒或超胞),可适当降低网格密度(如NGXF=150)以减少计算耗时,同时通过测试确保精度损失可控。
4. 从数据到洞见:Bader电荷的深度应用
掌握Bader电荷分析后,研究者可进一步将其与其他计算手段结合,实现多尺度、多维度的材料解析:
· 与态密度(DOS)联用:通过关联Bader电荷与特定轨道的态密度分布(如d带中心),揭示电荷转移的轨道贡献机制。
· 动态过程辅助分析:在分子动力学模拟中,定期输出电荷密度文件并分析Bader电荷演化,可追踪反应路径中的电子重构行为。
· 机器学习数据源:将Bader电荷作为特征输入机器学习模型,可加速催化剂或电极材料的高通量筛选。
结语
Bader电荷分析不仅是理论计算的“终点”,更是科学发现的“起点”。通过本教程,您已掌握从电荷密度计算到原子电荷提取的全链条技能。下一步,请将这一工具应用于您的具体研究问题,无论是揭示催化活性起源、优化电池电极材料,还是解析低维材料的界面效应,Bader电荷都将成为您探索电子世界的有力透镜。愿您在电荷的海洋中捕捉到启发的火花,推动科研工作的突破与创新!
找华算做计算👍专业靠谱省心又省时!
益于理论计算化学的快速发展,计算模拟在纳米材料研究中的运用日益广泛而深入。科研领域已经逐步形成了“精准制备-理论模拟-先进表征”的研究模式,而正是这种实验和计算模拟的联合佐证,更加增添了论文的可靠性和严谨性,往往能够得到更广泛的认可。
华算科技已向国内外1000多家高校/科研单位提供了超过50000项理论计算和测试表征服务,部分计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。
