

引言:能源材料的数字化革命
在碳中和与能源转型的全球浪潮中,能源材料的高效设计与发现成为科技突破的核心。传统实验方法受限于“试错”周期长、成本高,而计算材料学的兴起正通过“原子级模拟+数据驱动”颠覆这一领域。2024年《WIREs计算分子科学》的综述论文《能源相关材料的计算设计:从第一性原理到机器学习》系统梳理了四大计算范式。本文以“深度翻译+本土化解读”还原其核心框架,揭示计算材料学如何从理论走向实践。

1. 背景介绍
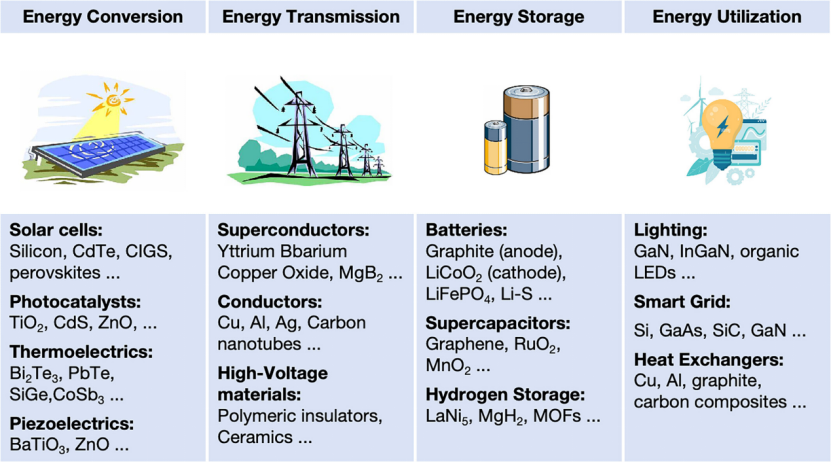
能源相关材料贯穿于能源生成、转换、传输、存储和利用的整个生命周期。这些材料包括用于光伏、电池、燃料电池、氢储存、热电、超导体以及能源转换催化剂等多种用途。它们对于推进能源技术、提高效率、降低成本以及减少环境影响至关重要。通过推进这些技术,能源相关材料对于满足全球能源需求和促进环境可持续性至关重要。
实验方法在开发新材料中起着关键作用,但传统上依赖于试错法,具有广泛的迭代周期和有限的候选评估。无机晶体结构数据库(ICSD)目前包含约10^5种实验验证的无机化合物,但与前103种元素的四组分化合物可能组合的10^12种相比,这只是几个数量级少。这种差异突出了当前实验探索的不完整性,并为材料发现的突破提供了重要机会。
计算材料设计,包括量子力学计算、分子模拟和数据驱动的机器学习技术,已经重塑了材料发现的研究范式。这些方法不仅阐明了材料特性,还实现了新材料的计算设计,显著扩展了已知材料,并产生了开放数据库,如材料项目(Materials Project)、开放量子材料数据库(OQMD)等。计算能力的进步和计算方法的发展使得筛选数千种候选材料以识别具有目标特性的材料成为可能,加速了新型能源相关材料的发现。
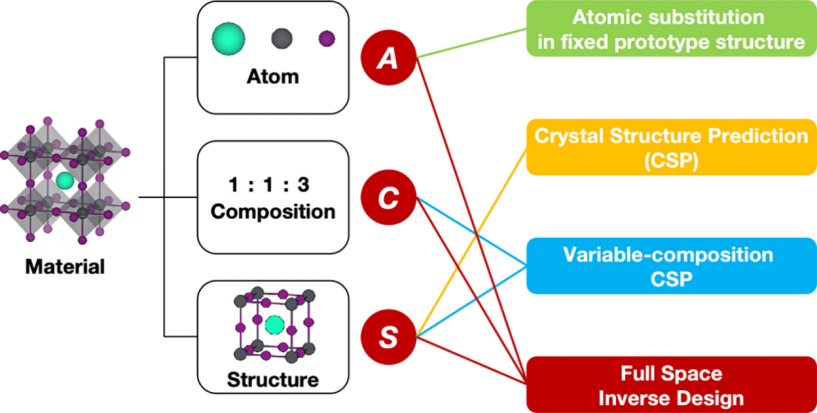
从计算材料设计的角度来看,材料由其构成原子(A)、成分(C)和结构(S)编码。具有不同ACS的材料表现出不同的电子、光学、机械和磁性特性。计算方法通过操纵这些ACS变量来识别具有所需特性的材料。然而,材料配置的完整空间(简称“全空间”)极其庞大,即使是简单的化学元素组合也会产生大量可能的化合物。盲目探索材料空间是一项艰巨的任务。因此,需要有效的策略来有效采样搜索空间。
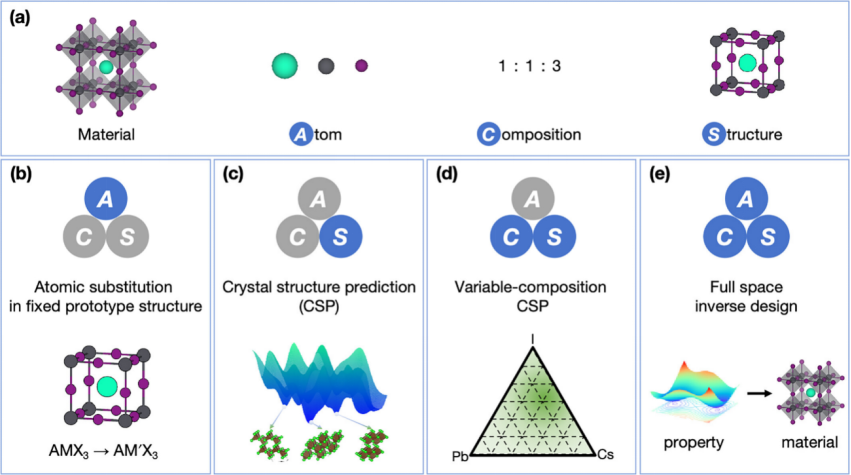
基于搜索空间中涉及的ACS维度,计算材料设计可以分为以下四类:
1.固定原型结构中的原子替换:在固定C和S的情况下搜索A空间。
2.晶体结构预测(CSP):在固定A和C的情况下搜索S空间。
3.变成分CSP:在固定A的情况下搜索C和S空间。
4.全空间逆向设计:在没有任何限制的情况下,根据目标特性预测材料的ACS。
2. 四大计算范式详解
范式一:固定原型结构中的原子替换
固定原型结构中的原子替换是一种高效的材料设计方法,它通过在已知结构框架中替换原子来探索新材料的性质。这种方法的核心思想是利用已知结构的稳定性和特性,通过替换其中的原子来实现材料性能的优化或新功能的开发。
1. 原子替换的基本原理
在固定原型结构中,原子替换的基本思路是保持材料的成分(C)和结构(S)不变,仅改变构成原子(A)。这种方法通常遵循以下两种模式:
·直接等价替换:用同一族周期表中的元素替换目标原子,例如将Zn(II)S替换为Cd(II)S。
·交叉替换或阳离子突变:用周期表中相邻元素替换目标原子,同时保持总价电子数不变,例如将Zn(II)S替换为Cu(I)Ga(III)S2。
这种方法的优势在于:
1.高效性:在已知结构框架内进行替换,减少了探索新材料空间的复杂性。
2.可预测性:基于已知结构的性质,可以预测替换后材料的性能。
3.实验可行性:替换后的材料更容易通过实验验证。
2. 基于第一性原理计算的原子替换
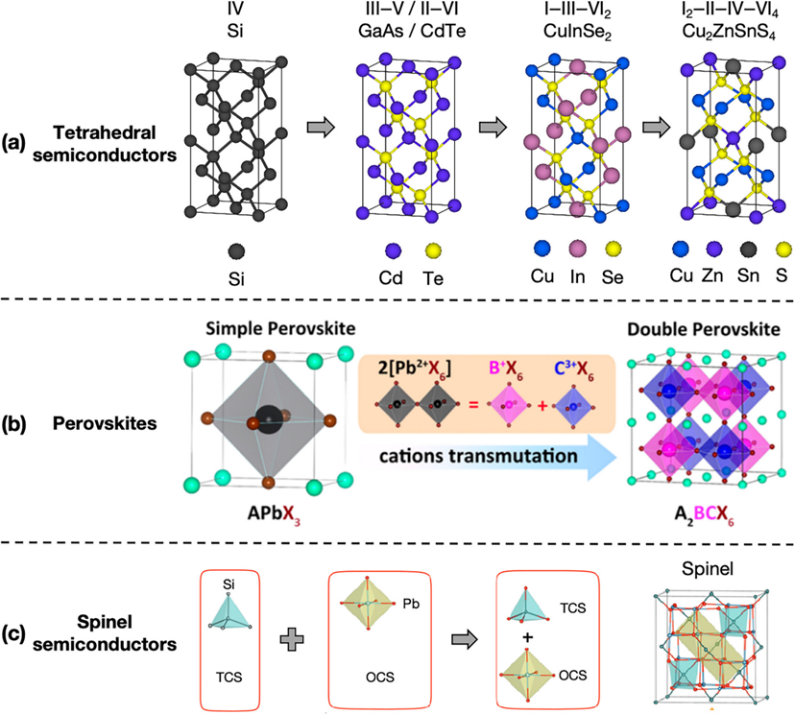
原子替换方法在半导体材料的发展中起到了重要作用。从硅到先进多元化合物的演变可以看作是一个原子替换的旅程。
·硅的钻石立方结构:硅的钻石立方结构是四面体配位的典型代表,每个硅原子被四个其他硅原子包围。这种结构为后续的III-V化合物(如GaAs)和II-VI化合物(如CdTe)提供了基础。
·III-V和II-VI化合物:通过替换硅结构中的原子,科学家们开发出了具有更高电子迁移率、直接带隙和强太阳吸收特性的半导体材料。
·钙钛矿结构:钙钛矿结构(如CH3NH3PbI3)因其优异的光电性能而备受关注。通过替换钙钛矿结构中的A+阳离子(如CH3NH3+替换为Cs+)或M2+阳离子(如Pb2+替换为Sn2+),可以设计出具有更高稳定性和环境友好性的新型钙钛矿材料。
3. 机器学习加速的原子替换
随着机器学习技术的发展,原子替换方法得到了进一步加速。机器学习模型可以快速预测材料的性质,从而显著减少计算成本。
·符号回归(Symbolic Regression, SR):通过数学公式将材料参数(如晶体结构、原子序数、离子半径等)与材料性能(如吸附能、d带中心等)关联起来。例如,Weng等人通过SR发现了一个简单的公式(μ/t,其中μ是八面体因子,t是容忍因子),可以线性关联过电位,从而快速筛选出具有高氧进化反应(OER)活性的钙钛矿氧化物催化剂。
·生成模型:生成模型(如变分自编码器VAE和生成对抗网络GAN)可以学习训练材料的分布,并基于此生成新材料。例如,iMatGen通过VAE编码晶体结构,成功生成了已知和新型的钒氧化物材料。
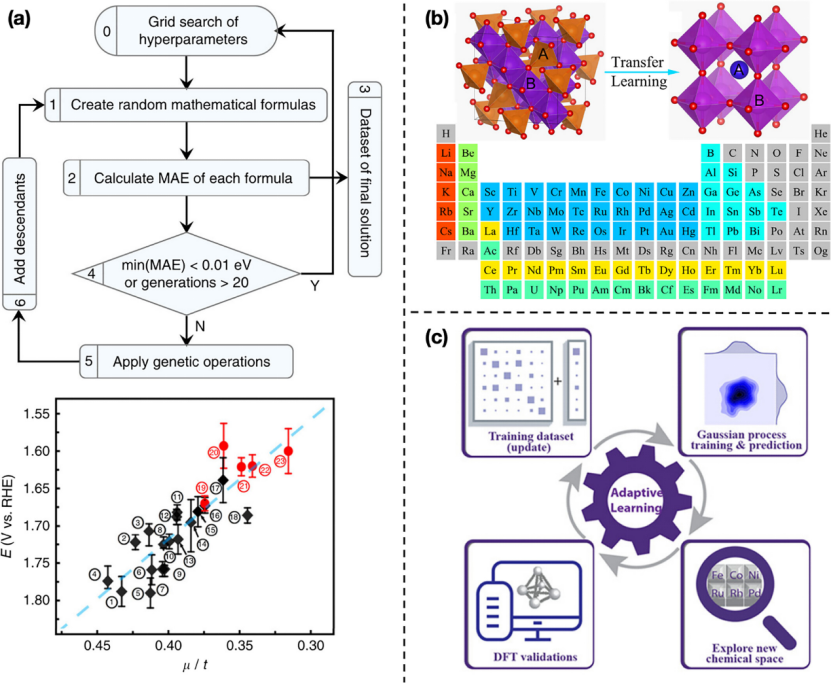
4. 应用案例
原子替换方法在能源相关材料的设计中取得了许多重要成果:
1.钙钛矿太阳能电池:通过替换钙钛矿结构中的A+阳离子(如CH3NH3+替换为Cs+)或M2+阳离子(如Pb2+替换为Sn2+),开发出了更稳定、更环保的钙钛矿太阳能电池。
2.双钙钛矿:通过阳离子突变设计出了无铅的双钙钛矿(如BaZrS3),这些材料在保持优异光电性能的同时,避免了铅的毒性问题。
3.传统和新型半导体:通过替换传统半导体(如CdTe)中的原子,开发出了具有更高稳定性和效率的新型半导体材料。
4.二维材料:通过替换石墨烯中的碳原子(如用硼和氮替换两个碳原子形成h-BN),设计出了具有优异电子性能的二维材料。
范式二:晶体结构预测(CSP)——解锁材料的“能量密码”
1. 基本原理与核心目标
晶体结构预测(Crystal Structure Prediction, CSP)的核心任务是:在给定化学组分(元素种类与配比固定)的条件下,通过计算寻找能量最低的晶体结构。材料性能(如电子能带、光学吸收、离子导电性)对原子排列高度敏感。例如:
·碳的同素异形体:金刚石(绝缘体)、石墨(导体)、石墨烯(半金属)性能迥异。
·CsPbI₃钙钛矿:不同晶相(α、β、γ、δ)的带隙与稳定性差异显著,直接影响光伏效率。
CSP的输入仅为原子种类和数量,输出为全局能量最低的稳定结构及可能的亚稳态结构。其本质是在超高维势能面上搜索能量最小值,涉及两个关键环节:
1.能量计算:通过量子力学方法(如DFT)或机器学习势函数(MLP)评估结构能量。
2.优化算法:采用智能搜索策略(如遗传算法、粒子群优化)高效遍历构型空间。
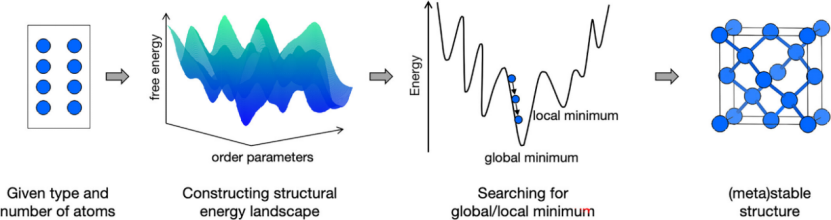
2. 算法演进:从DFT到机器学习势函数
传统DFT驱动的CSP
早期的晶体结构预测(CSP)方法高度依赖密度泛函理论(DFT)进行能量计算,结合全局优化算法实现结构搜索。其核心流程可分为三个步骤:
1.初始结构生成:基于对称性约束或完全随机方式,生成大量候选晶体结构。例如,通过随机堆砌原子或依据已知晶体原型(如钙钛矿、尖晶石)构建初始模型。
2.DFT结构弛豫:利用DFT对每个候选结构进行几何优化,计算其总能、原子受力与应力,迭代调整原子坐标与晶胞参数直至收敛。
3.低能构型筛选:通过能量排序或热力学稳定性分析(如凸壳计算),筛选出全局能量最低的稳定结构及可能的亚稳态构型。

典型的优化算法包括:
·进化算法(如USPEX):模拟生物进化机制,通过“变异”(随机调整原子位置)、“交叉”(交换不同结构的片段)和“选择”(保留低能个体)逐代优化种群。例如,USPEX在高压氢化物预测中成功发现了H₃S超导相。
·粒子群优化(如CALYPSO):模拟鸟群协同搜索行为,通过共享群体最优解信息加速收敛。CALYPSO在硅基材料设计中预测出20多种新型硅同素异形体。
·随机采样与盆地跳跃(Basin Hopping):通过蒙特卡洛随机扰动结合局部优化,跨越势能面能垒以探索全局最小值。
然而,传统DFT-CSP存在显著瓶颈:
·计算成本高昂:单次DFT弛豫需数小时至数天(如100原子体系在普通计算集群需10-20小时),导致百万级结构筛选难以实现。
·体系规模受限:复杂组分(如三元以上化合物)或大晶胞(>200原子)的计算资源需求呈指数增长。
·亚稳态预测困难:依赖静态能量计算,难以评估动力学稳定性(如势垒高度与寿命)。
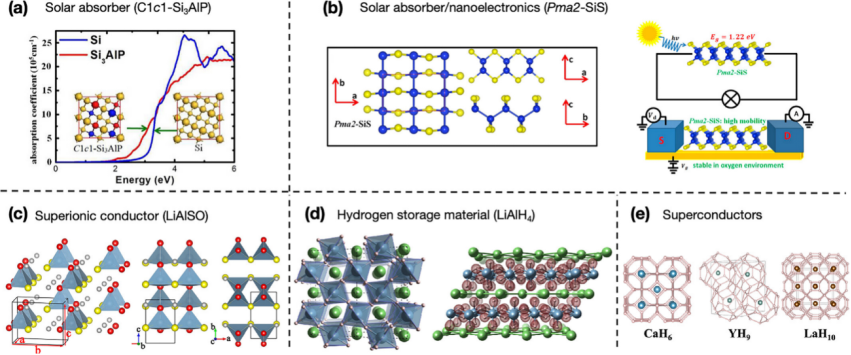
范式三:可变成分晶体结构预测
1. 核心定义与目标
可变成分晶体结构预测(Variable-Composition CSP)旨在固定元素种类(如B-C-N体系),探索所有可能的化学配比(成分C)及其对应的稳定晶体结构(结构S)。与传统CSP(固定成分)不同,它需同时优化成分与结构的双重变量,目标包括:
·全局相图构建:确定热力学稳定相及其稳定区间(如压力、温度条件)。
·隐藏化合物发现:揭示实验未报道但理论上稳定的新材料。
·性能导向设计:筛选具有目标特性(如高硬度、超导性)的成分–结构组合。
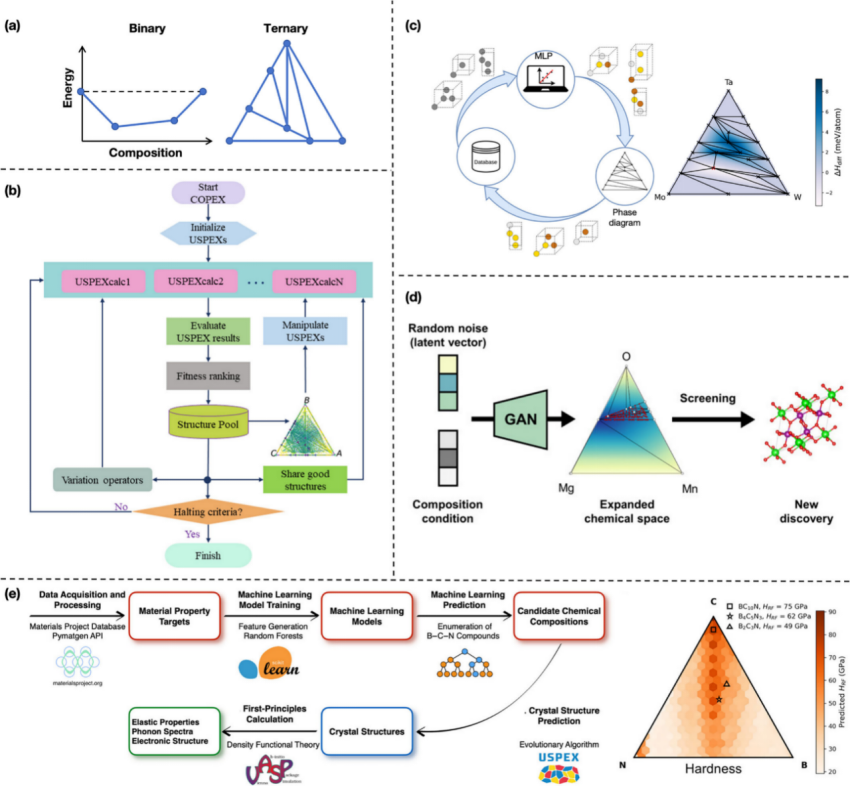
范式四:全材料空间反向设计
目标:不预设元素、成分与结构,直接从目标性能反向推导材料。
·三大策略:
高通量筛选(HTS):从数据库(如Materials Project)筛选数万种化合物,发现43种新型CO₂光催化剂。
全局优化:全空间逆设计框架(FSIMD)结合元素增强采样(EES),高效定位高性能材料。
生成模型:
MatterGen:基于扩散模型生成满足对称性、稳定性的全新结构,成功率提升15倍。
FTCP框架:通过傅里叶变换晶体表征,设计出热电性能媲美GeTe的新材料Au₂Sc₂O₃。
未来:生成模型需突破“仅限稳定性”的约束,融入更多性能指标。
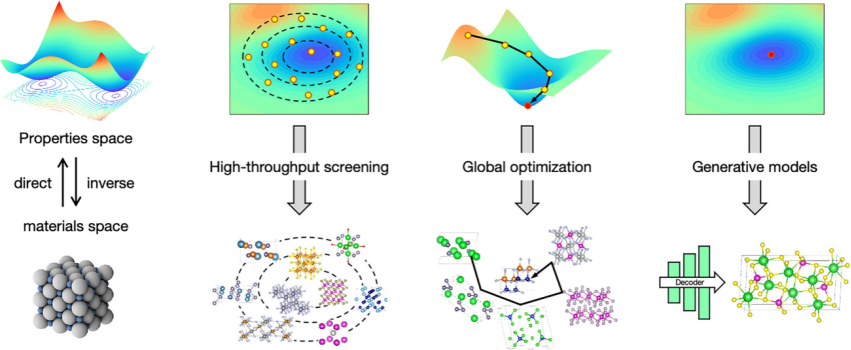
高通量筛选(HTS)
HTS是全材料空间逆向设计的最早尝试。这种方法从基于研究人员直觉创建的初始候选材料库开始,通常数量从10^3到10^5不等。然后根据预定义的目标特性(如稳定性、电子和光学特性以及合成的难易程度)对初始候选材料进行过滤。HTS在设计高性能能源相关材料方面取得了显著成功。
全局优化
全局优化通过使用准随机搜索、模拟退火、盆地跳跃、元动力学、最小值跳跃、遗传算法、进化算法、蒙特卡洛树搜索(MTCS)算法等方法,高效地探索巨大的材料空间,以识别具有目标特性的材料。
生成模型
生成模型是逆向设计的新兴前沿方法。它们通过学习训练材料相对于目标特性的分布来生成新材料。这些模型将高维化学空间编码为低维连续向量空间(潜在空间),从而简化了对复杂、高维材料空间的探索。

3. 结论与展望
计算材料设计在加速新型能源相关材料的设计和发现方面发挥了重要作用。为了高效探索由ACS变量编码的巨大材料空间,计算材料设计范式被分为固定原型晶体结构中的原子替换、CSP、变组成CSP以及全材料空间中的逆向设计。尽管机器学习辅助的材料设计在效率和效果上取得了显著进展,但仍存在一些挑战,包括确定决定性能的关键物理和化学特性,以及找到与宏观特性相关的可计算参数。此外,开发能够预测晶体可合成性的机器学习模型也是一个重要挑战。
论文链接:https://wires.onlinelibrary.wiley.com/doi/abs/10.1002/wcms.1732
找华算做计算👍专业靠谱省心又省时!
益于理论计算化学的快速发展,计算模拟在纳米材料研究中的运用日益广泛而深入。科研领域已经逐步形成了“精准制备-理论模拟-先进表征”的研究模式,而正是这种实验和计算模拟的联合佐证,更加增添了论文的可靠性和严谨性,往往能够得到更广泛的认可。
华算科技已向国内外1000多家高校/科研单位提供了超过50000项理论计算和测试表征服务,部分计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。
