

VASP计算差分电荷的具体流程如下:
1. 优化结构:首先需要优化整个系统的结构,例如OOH吸附在NiO(111)表面的情况。这一步确保了三次自洽计算中使用的FFT网格一致性(NGXF, NGYF, NGZF)。
2. 分别优化片段:然后分别优化系统中的各个片段,例如OOH和NiO(111)表面,以获得各自的CHGCAR文件,这些文件包含了电荷密度信息。
3. 计算差分电荷密度:使用vtstscript脚本包进行电荷差分计算,该脚本可以读取不同CHGCAR文件并计算电荷密度差,得到差分电荷文件。
4. 可视化:将生成的差分电荷文件导入到VESTA软件中进行可视化,以观察电荷密度的变化。在VESTA中,可以通过设置等值面来显示电荷密度的增加或减少。
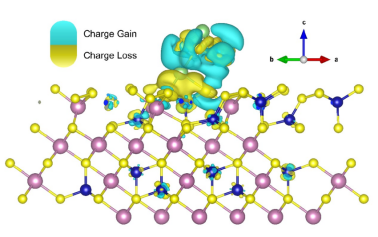
通过这些步骤,可以分析成键过程、结构弛豫前后的电荷转移,以及吸附分子与衬底之间的电荷传输等。
晶体结构优化结果:


差分电荷计算结果:
自洽计算INCAR文件:

声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!