



第一作者和单位:刘焜 南昌大学
通讯作者和单位:廖光福 福建农林大学
原文链接:https://doi.org/10.1016/j.ccr.2025.216611
二氧化碳高效转化为高附加值轻质烯烃(C2-C4=)具有重要工业价值,既能生产关键化工原料,又能减少温室气体排放。尽管二氧化碳加氢技术取得显著进展,但开发能高效活化C-O键、促进C-C链增长并实现低碳烯烃高选择性的催化剂仍是核心挑战。本文聚焦铁基催化剂改性的费托合成技术,该技术具有能量效率高、适合通过热催化二氧化碳加氢生产C2-C4=的优势。重点解析催化活性铁物种的动态特性、新型材料创新及突破Anderson-Schulz-Flory(ASF)分布限制与低烯烃选择性的先进催化剂设计策略。通过系统阐述反应机理、催化剂组成与性能调控要素,为下一代高效可持续催化系统开发提供理论指导,搭建基础研究与前沿技术的转化桥梁。
近年来,CO2过量排放引发的环境问题日益严峻。通过将CO2转化为烃类或含氧化合物等化学品,不仅可有效缓解碳排放压力,更能实现环境保护与经济可持续发展的双重目标。低碳烯烃(乙烯、丙烯、丁烯)作为生产溶剂、医药、聚合物的重要中间体,其传统制备工艺(石脑油制烯烃、煤制烯烃、丙烷脱氢制烯烃)存在高能耗、不可持续等问题。因此,CO2加氢制烯烃技术成为研究热点,其规模化应用既能创造经济效益,又能实现碳循环闭环,对构建清洁能源产业链具有重要意义。
CO2制烯烃主要有两条技术路径:一是通过逆水煤气变(RWGS)与费托合成(FTS)耦合的CO2-FT路线;二是经甲醇中间体的甲醇制烯烃(MTO)路线。其中,CO2-FT路线需催化剂同时具备RWGS和FTS双功能活性。由于RWGS为吸热反应,高温条件(>523 K)有利于CO生成,但铁基催化剂在该过程中面临诸多挑战:一方面,传统费托合成遵循ASF分布规律,C2-C4选择性通常低于60%,且甲烷选择性偏高,导致产物分离成本增加;另一方面,铁基催化剂易积碳烧结失活,再生工艺制约其工业化应用。提升铁基催化剂性能的关键在于:(1)精准调控活性铁物相(Fe3O4、χ-Fe5C2、ε-Fe2C等)及其动态演变;(2)通过助剂改性(碱金属、过渡金属等)、载体优化(碳基材料、分子筛等)或新型结构设计(核壳结构、金属/分子筛双功能体系等)调控表面电子特性;(3)优化制备工艺(水热法、溶胶–凝胶法等)与反应条件(温度、CO2/H2比)。本文系统综述铁基催化剂在CO2加氢制烯烃领域的研究进展,重点剖析活性位点特性、失活机制与反应路径,为设计高效稳定催化剂提供理论指导。
铁相物种
铁碳间化合物作为费托合成(FTS)关键催化材料,其晶相结构(χ-Fe5C2、ε-Fe2C/Fe2.2C、θ-Fe3C、Fe7C3)对催化性能具有决定性影响(图1)。尽管历经近百年研究,活性位点与优势晶相的争议仍未平息。χ-Fe5C2相作为经典活性相,通过Na-Zn共修饰可调控表面电子态,抑制烯烃过度加氢,使C5+烯烃选择性提升40%。ε-Fe2C/Fe2.2C相在低温FTS中表现突出,通过优化碳化参数可制备超低CO2选择性催化剂,在235 ℃下稳定运行150小时。θ-Fe3C相传统被视为惰性相,但Mn掺杂实验揭示其碳缺陷结构可高效催化轻质烯烃生成,选择性达60.1%。Fe7C3相虽研究较少,但在高碳势环境下展现极高本征活性(TOF=4.59×10⁻² s⁻¹)。晶相调控策略呈现多元化趋势:MOF衍生法可实现相结构温度依赖性调控;环境透射电镜直接观测相变过程;合金碳化法可定向合成优势相。值得注意的是,初始氧化铁晶型(α-Fe2O3vs γ-Fe2O3)显著影响碳化路径,γ-Fe2O3基底更易形成活性θ-Fe3C相。当前挑战集中于多相共存体系的活性位辨析、相稳定性与产物选择性的平衡机制。未来研究需结合原位表征与理论计算,深化对动态相变过程的理解,以突破传统经验调控模式,实现催化体系的理性设计。
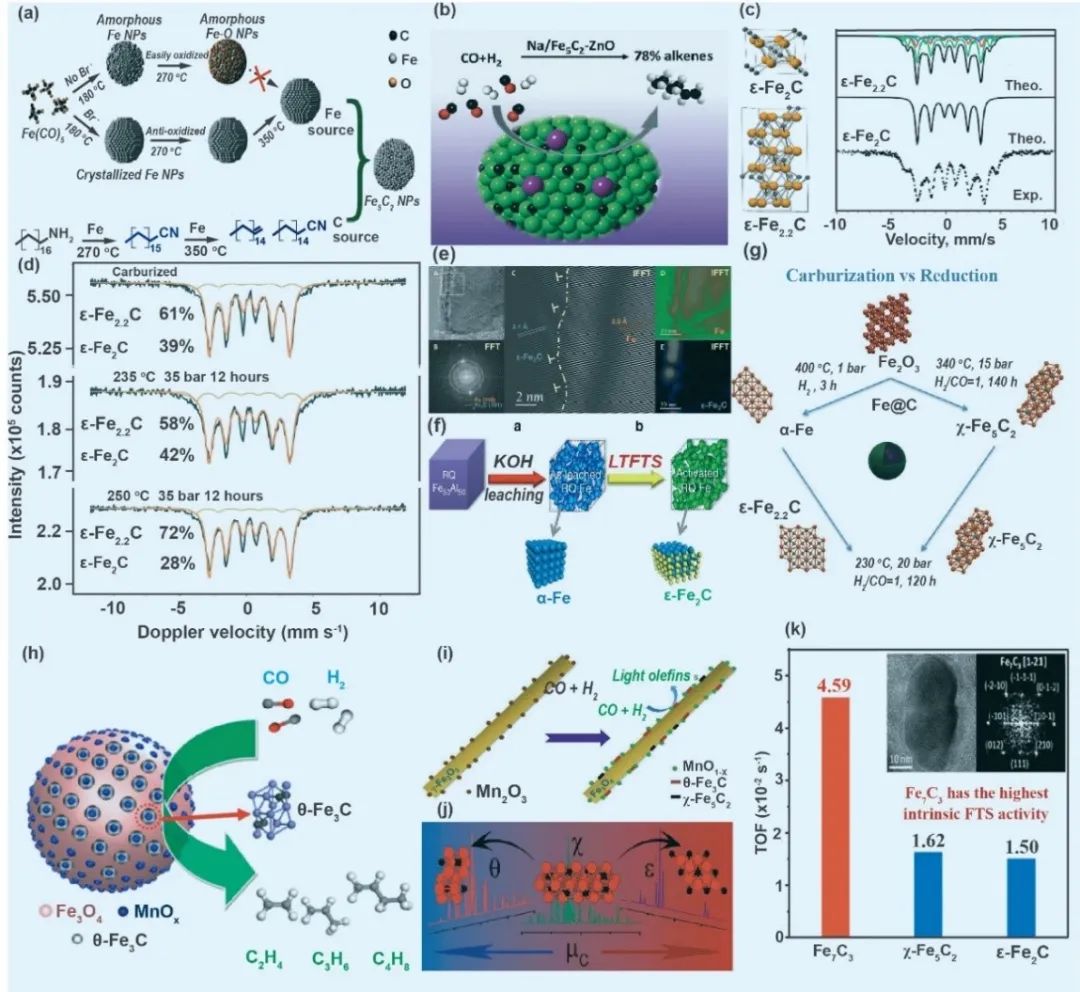
图1 铁碳间化合物作为费托合成(FTS)关键催化材料,其晶相结构(χ-Fe5C2、ε-Fe2C/Fe2.2C、θ-Fe3C、Fe7C3)对催化性能具有决定性影响的一些案例
铁相物种转变
将二氧化碳加氢转化为C2+烃类的核心技术在于耦合逆水煤气变换反应(RWGS)与费托合成(FTS),通过改性FTS催化剂实现烯烃或烷烃生产。铁基催化剂通过Fe3O4与Fe5C2的协同作用,分别催化RWGS和FTS反应。当前研究通过添加助剂、载体或结构设计优化催化剂,调控表面电子态和铁相平衡以提升性能。反应过程中铁物种呈现多相演变:Fe3O4主导RWGS,金属铁和碳化铁促进CO加氢生成烃类(图2)。温度与H2/CO比显著影响碳化铁相变:高温(>623K)及低碳化学势(高H2/CO比)促进θ-Fe3C生成,低温(523-623K)形成χ-Fe5C2,温度超过723K时χ-Fe5C2完全转化为θ-Fe3C。低温下()渗碳生成ε-Fe2C系列碳化物,FTS典型条件(>523K)下进一步转化为χ-Fe5C2。关于活性位点仍存在争议:Fe3O4被认为是RWGS活性相,Fe5C2催化FTS反应,但还原过程中铁相从氧化物向金属态转变,并在CO环境中形成碳化物。Riedel等提出稳态反应动力学五阶段模型:初始吸附活化→碳沉积引发RWGS主导→FTS活性稳定→最终表面重构。催化剂失活主要源于水生成导致的碳沉积,但氢还原产生的氧空位可激活CO2生成CO中间体。铁氧化物前驱体经H2/CO还原形成金属铁或碳化铁,二者均具备CO解离与加氢能力,其中FTS过程尤其促进碳化铁形成。
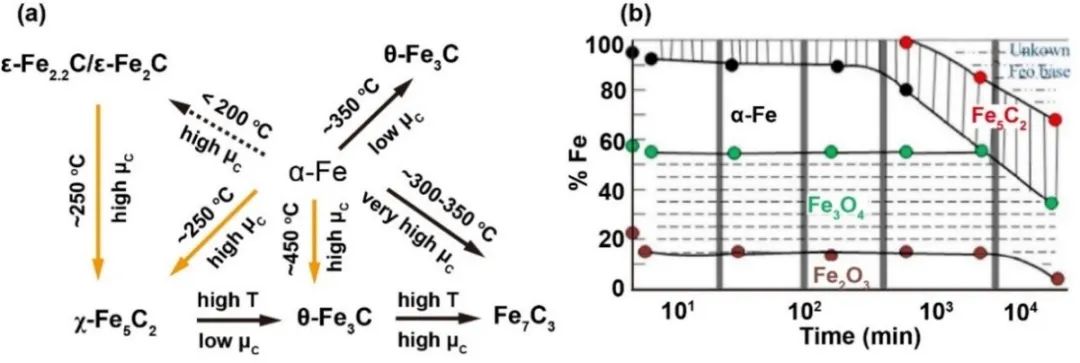
图2 铁物相相互转变的示意图
铁相与催化活性的关系
铁基催化剂的物相演变显著影响其反应活性(图3)。反应前催化剂需在H2中预还原,形成Fe3O4、FeO或金属铁(Fe⁰)。在反应气氛中,这些物相逐步发生渗碳,转化为χ-Fe5C2、θ-Fe3C和Fe3C7。反应过程中催化剂体系呈现多相共存(α-Fe、α-Fe2O3、Fe3O4及各类铁碳化物)。研究证实Fe3O4是水煤气变换逆反应(RWGS)的活性相,而χ-Fe5C2在费托合成(FTS)中起关键作用。
碳化铁的结构特性直接影响催化性能:χ-Fe5C2的氢解能力弱于θ-Fe3C,导致烯烃二次加氢概率降低,从而提升低碳烯烃选择性(O/P比升高);θ-Fe3C因强CO2吸附促进C-C偶联,C5+产物选择性更高。但单纯χ-Fe5C2催化时,CH4(45%)和低碳烷烃(25%)仍为主产物,需优化选择性。钾助剂(K)可加速α-Fe向χ-Fe5C2转化,通过促进CO解离提高渗碳速率和χ-Fe5C2丰度,但过量K可能削弱效果。制备温度显著影响物相:低温(593K)草酸盐热解可获高χ-Fe5C2含量样品,展现优异轻烯烃选择性;高温(1173K)则促进θ-Fe3C生成,链增长能力增强(α=0.55)。原位表征确认Fe5C2为烯烃生成活性相,其不可逆氧化是催化剂失活主因。还原–渗碳过程遵循α-Fe2O3→Fe3O4→Fe→χ-Fe5C2的相变序列,Fe(II)物种为碳化主要前驱体,最终建立碳化物与Fe(II)氧化物的动态平衡。
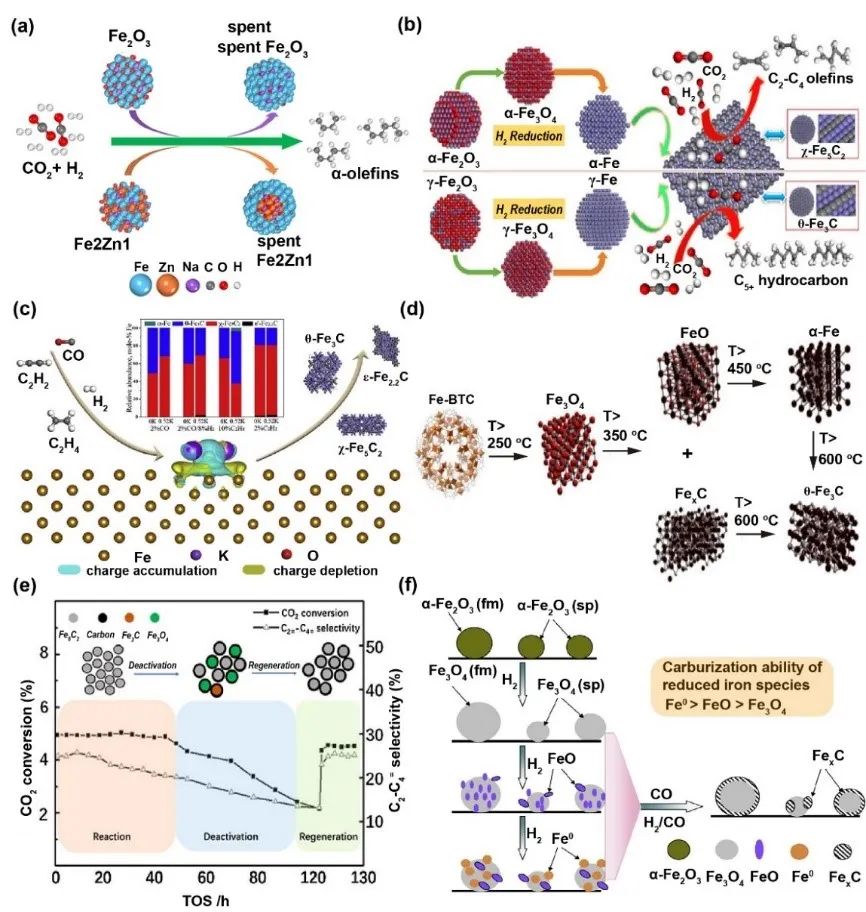
图3 铁物相与催化活性的关联的一些案例
Fe活性相的失活
铁基催化剂在费托合成(FTS)过程中面临多机制失活,主要包括相变、烧结、碳沉积和中毒(图4)。活性Fe物种(如Fe5C2、χ-碳化物等)在CO2/H2反应气氛中易氧化为Fe3O4,导致催化性能丧失。研究证实,富氧原料气中CO2和H2O作为氧化剂加速相变,而烧结现象则因铁物种迁移聚合造成比表面积下降。研究发现,含Fe3O4的失活催化剂可通过10% CO在350 ℃下处理5小时再生Fe5C2,而Fe3C需两步法(CO2-CO氧化–渗碳)恢复活性。反应副产物H2O对催化剂结构具有显著破坏作用。其氧化特性促使Fe物种向Fe2O3转化,同时改变催化剂孔隙结构。研究表明,当反应器出口水氢比(pH2O/pH2)超过阈值时,催化性能急剧下降。研究表明在降低水进料比条件下实现CO2转化率和液体燃料产率双提升,但过度脱水反而导致活性衰减。
总之,铁基催化剂失活涉及氧化相变、结构破坏和碳堵塞多因素协同作用。通过精准调控反应气氛、优化催化剂组分及开发再生工艺,可有效延长催化剂寿命。未来需重点关注水管理策略与原位再生技术开发,以突破CO2加氢制燃料技术的工业化瓶颈。
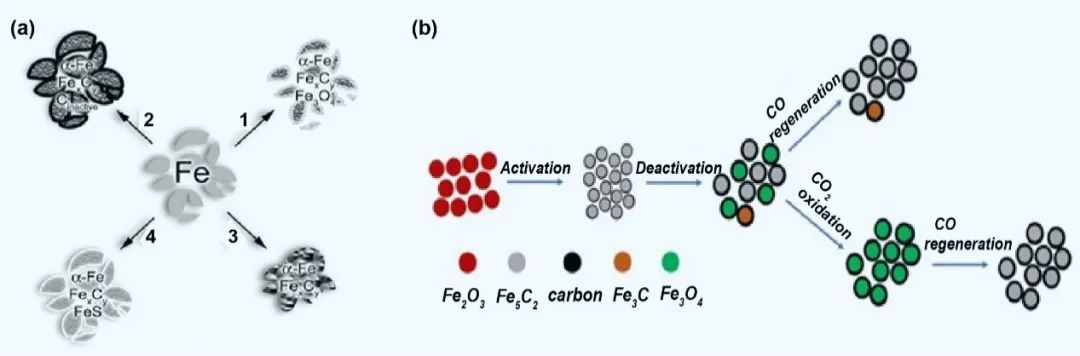
图4 铁物相与催化活性的关联的一些案例
其它活性相
当前研究表明,Co、Ru、Ni、Fe等金属在CO2加氢制低碳烷烃反应中表现出显著活性差异(图5)。不同催化剂的产物分布具有明确特征:
1. 钴基催化剂广泛用于费托合成(FTS)及CO2制烃过程,对碳链增长反应活性较高,但对水煤气变换(WGS)及逆水煤气变换(RWGS)反应活性较弱。其性能易受原料碳氢比和温度影响,RWGS活性低于铁基催化剂,甲烷副产物生成量较大。
2. 镍基催化剂在CO2甲烷化反应中表现突出,研究表明,高度分散的Ni/La2O3催化剂在208-380 ℃范围内可实现100%甲烷选择性,且CO2转化率随温度升高持续提升。但该催化剂不适用于低碳烃合成。
3. 钌基催化剂催化性能卓越,但高昂成本和资源稀缺性限制其工业应用。热力学研究表明,Ru/Al2O3催化剂的优化分散可降低反应活化能,但主要产物为甲酸(HCOOH)。
铁基催化剂兼具低成本与RWGS/FTS高效性,在CO2制低碳烯烃领域展现出独特优势。其应用灵活性高于其他金属,尤其适合需兼顾RWGS活性和产物调控的场景。
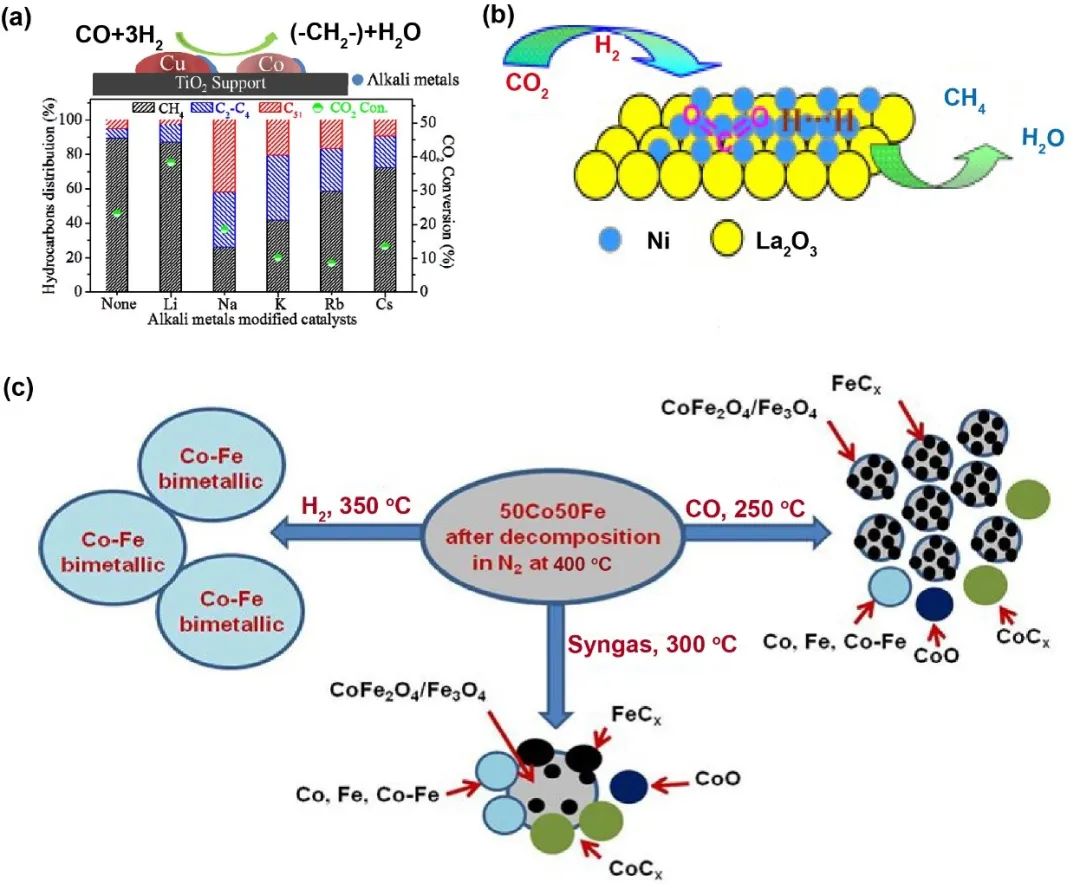
图5 其它活性相(非铁)以及特点的一些案例
反应微环境中催化剂相的调节
在CO2加氢反应中,Fe5C2作为铁基催化剂的高效助催化剂,通过结构调控可显著影响产物分布(图6)。还原气氛(H2、CO、CO/H2)中C/H/O比例变化会改变Fe5C2生成路径:H2气氛下遵循Fe2O3→Fe3O4→Fe→Fe5C2的还原碳化路径,而CO及CO/H2气氛中则直接生成Fe5C2。反应环境改变会导致Fe5C2颗粒尺寸及Fe5C2/FeOx比例动态变化,进而影响产物分布。CO2加氢过程伴随大量水生成,其表面分压会氧化活性铁碳化合物并抑制CO2活化,导致实际转化率(40-45%)低于热力学平衡预测值(>60%)。通过疏水壳层调控催化剂结构,可加速水分子脱附,降低表面水分压,促进RWGS反应向CO2消耗方向进行。研究表明,疏水壳层虽能提升H2O扩散,但DFT计算显示表面吸附水会提高C-C偶联能垒,促进CH₄生成而非碳链增长。研究发现,随着水含量从0%增至40%,CO2转化率降至20%,CO选择性降至6%,而FTS反应中CO转化率仅降至84%,表明水对CO2加氢影响更为显著。疏水层引入虽改善传质,但其对反应物扩散的阻碍作用及水对活化能的具体影响仍需深入研究。
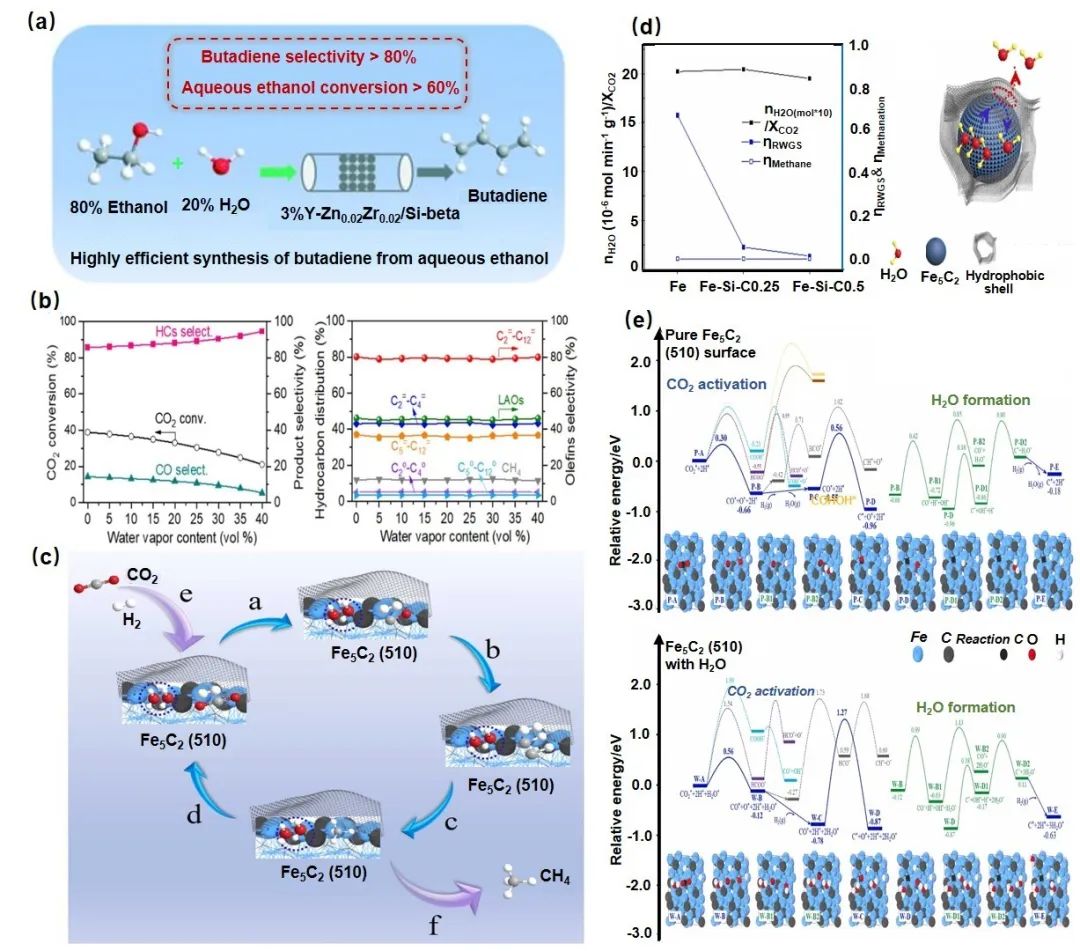
图6 反应微环境中催化剂相的调节对催化反应的影响的一些案例
控制氢碳比(H/C)对生产轻质烯烃至关重要,过量氢气可能导致加氢和甲烷化,而氢气不足会阻碍CO2转化。关键阶段包括断裂C-O键和生成C-C键。铁基催化剂在CO2费托合成(FTS)中表现优异,Fe3O4是主要活性成分。反应路径如图7a所示:RWGS催化剂活性位点(如Fe3O4)捕获并活化CO2,形成吸附的羧酸盐中间体(CO2),随后与吸附氢反应,生成HOCO中间体,进一步解离为OH和CO。OH加氢生成H2O,CO可脱附为气态CO或进入FTS步骤。对于烃类生成,CO部分加氢形成HCO,经完全加氢、解离和脱水,生成CHx物种,作为烯烃前驱体。CO也可解离为C和O片段,C可渗入铁晶格形成碳化铁(如Fe5C2),促进FTS反应。Fe5C2表面,C加氢生成CHx物种。C + CHx和CHx+ CHx*耦合是形成烯烃的主要路径。FTS中,解离的CO可渗入α-Fe晶格,形成Fe7C3、Fe5C2、θ-Fe3C等碳化铁,影响CO加氢、解离和C-C耦合。Fe5C2被认为是主要活性相,而Fe3C活性较低,可能产生石墨导致催化剂失活。甲醇合成有两种机制:CO依赖途径(通过RWGS反应生成的CO加氢,经HCO和COH中间体生成甲醇)和甲酸中心途径(羧酸盐加氢生成甲酸盐,经H2COOH、H2CO、H2COH和H3CO生成甲醇)。甲醇通过脱水耦合生成*CH2CH,再加氢生成烯烃。改性费托合成(MFTS)途径结合CO₂催化加氢和FTS步骤,需催化剂对RWGS和FTS均有活性。FeK/Al2O3催化剂主要遵循氧化还原机制。铁基材料在MFTS中用于烯烃生产,碳化物机制是主要烃类生成路径。整个CO2加氢反应机制如图7b所示,涉及CO2吸附、氧化、H分裂、中间体生成、碳链增长和终止步骤,铁基活性位点与CO2间发生氧化还原反应,通过碳化物介导机制实现链增长。
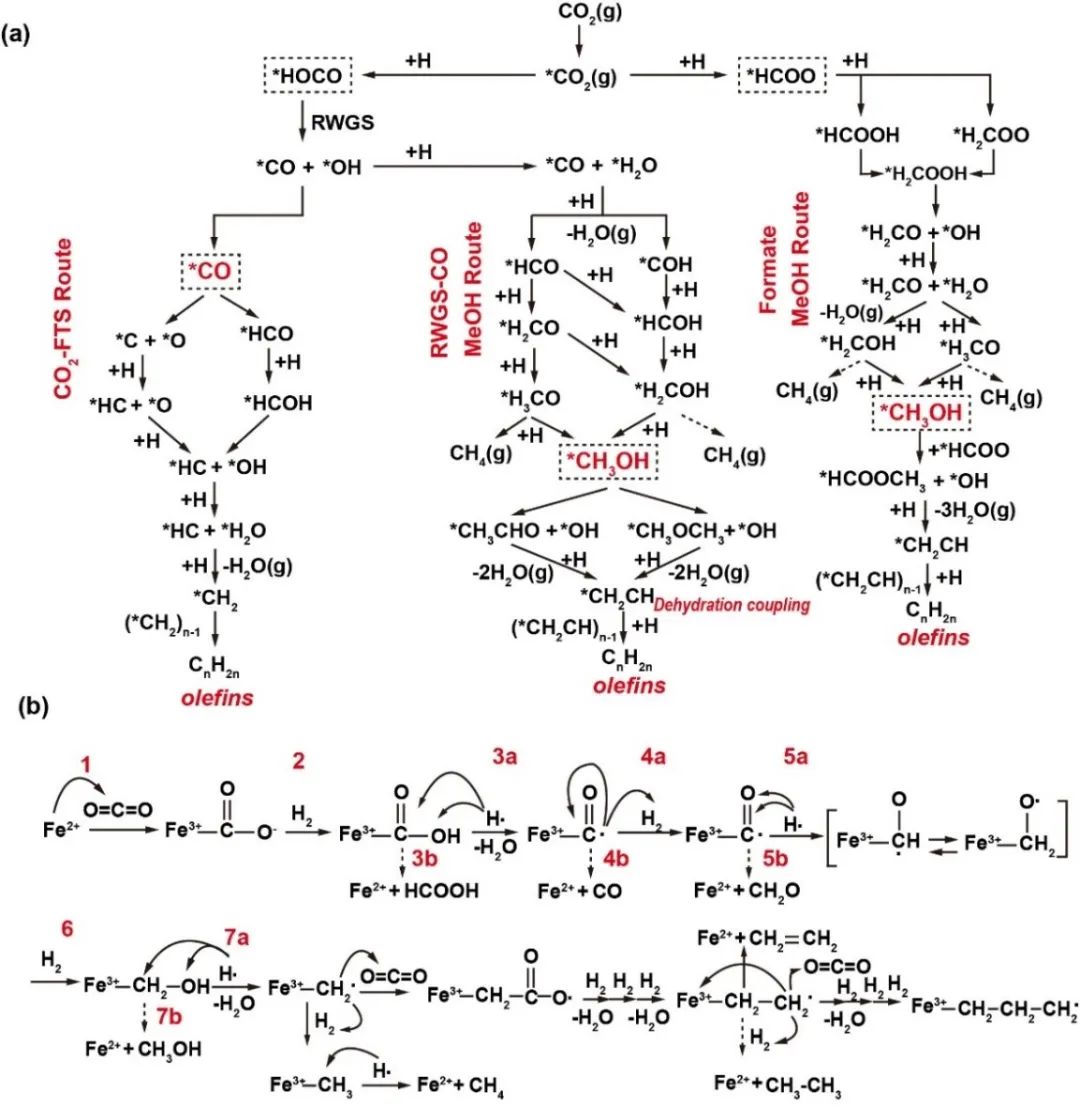
图7 反应微环境中催化剂相的调节对催化反应的影响的一些案例
引入助剂
Na-Fe
钠在CO2-FT反应中的多重作用机制被系统揭示(图8):其通过细化Fe5C2颗粒、提升表面钠密度,增强铁物种电子云密度以促进烯烃脱附;同时诱导碳沉积形成稳定FeCx物种,抑制中间产物过度加氢,显著提升烯烃/烷烃比。研究证实钠修饰可优化Fe5C2活性相的稳定性,并通过调节表面CH/CH2比例促进C-C耦合。多组实验表明,适量钠添加使CO2转化率及烯烃选择性分别提升至36.8%和64.3%,过量则趋近饱和。机理研究表明,钠通过电子转移降低氧空位形成能,削弱催化剂加氢能力,同时增强CO吸附并稳定短链烷基中间体,从而定向调控产物分布。碱金属碱性增强时,CO2吸附活化效率提升,进一步强化了低碳烯烃选择性。
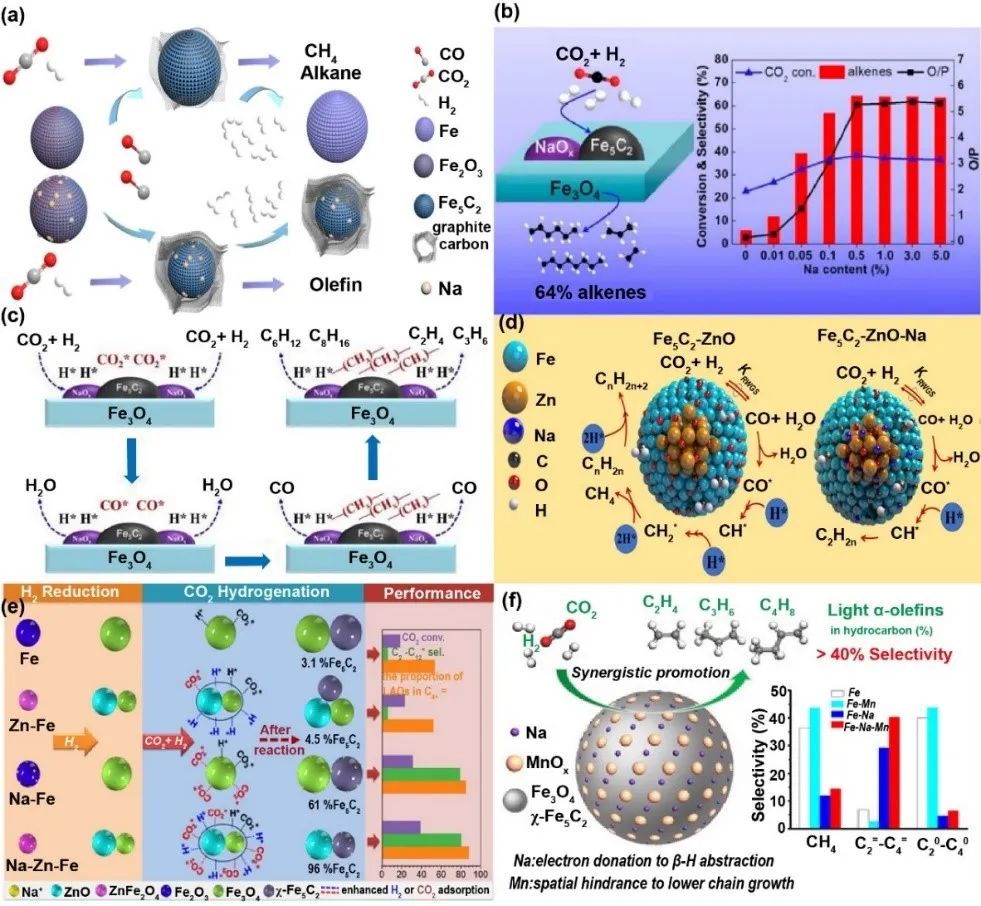
图8 Na助剂用于提升Fe基催化剂的一些案例
K-Fe
K2O在铁基费托合成催化剂中具有双重作用:初期通过抑制甲烷生成、促进碳链增长和铁碳化物形成,显著提高烯烃及含氧化合物选择性,但过量会导致表面积下降和活性衰减。研究表明,K⁺修饰通过降低H2吸附、增强CO2吸附,抑制低碳烯烃过度加氢,使烯烃选择性提升至44%且产率达11-13%。钾的给电子特性促进CO中间体吸附解离,加速碳沉积与Hägg碳化物(χ-Fe5C2)生成,优化铁活性相平衡。密度泛函理论(DFT)分析揭示,催化剂表面形成的K2CO3、KHCO3等物种在反应条件下可相互转化,当Fe/K比为2时,CO2转化率达44.2%,C2-C10烯烃选择性66.2%,其中53.3%为C2-C4优势产物。适量钾通过调控CO2-CO转化路径,协同铁氧化物与碳化物活性位点,实现高效费托合成。
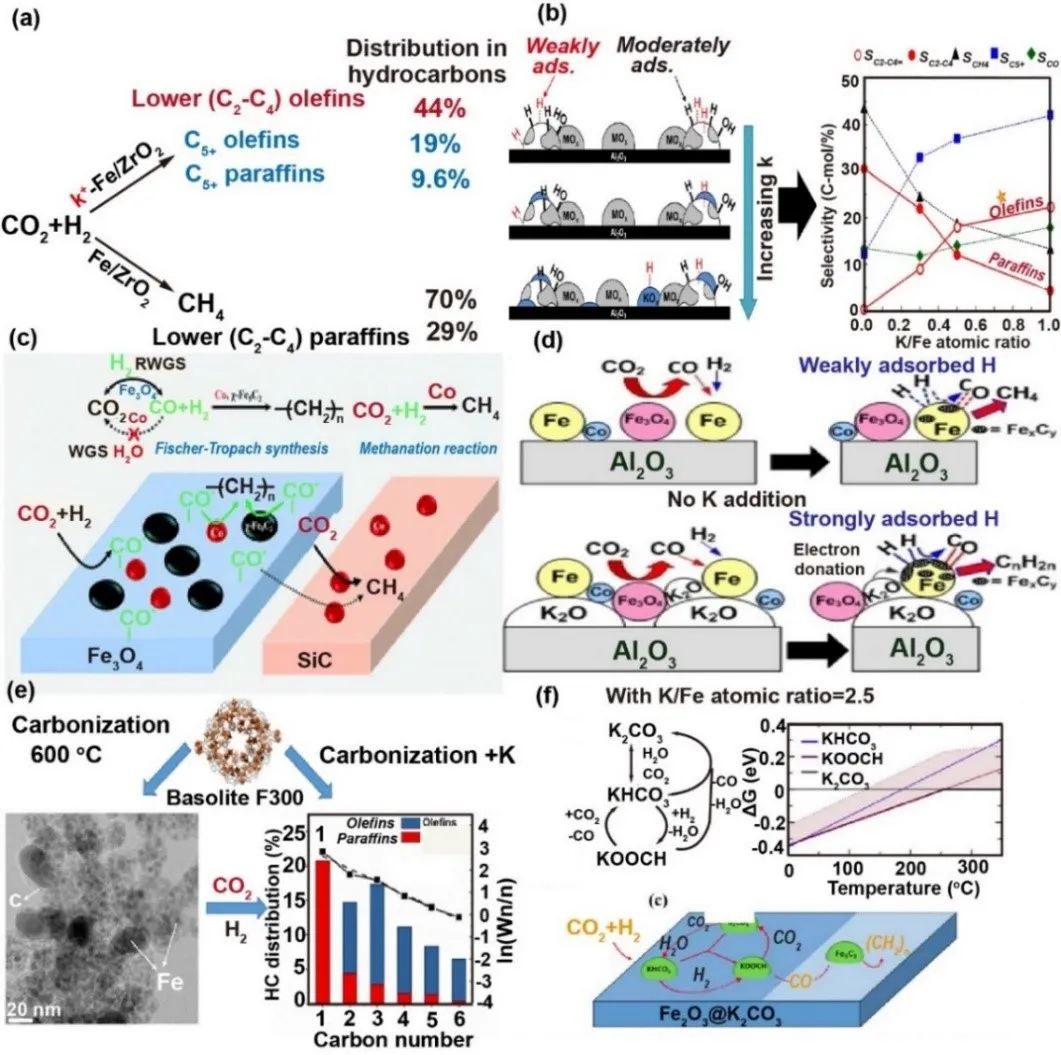
图9 K助剂用于提升Fe基催化剂的一些案例
载体材料对CO2制烯烃过程的影响
选择合适的载体能显著提升铁基催化剂的性能(图10)。载体通过调控表面pH、电子密度及孔结构,优化了催化性能与稳定性。碱性位点和氧空位是载体材料的独特优势,铁物种的还原碳化效率高度依赖金属与载体的相互作用强度。氧化物载体如SiO2、Al2O3等,能有效抑制活性中心烧结,促进催化剂还原碳化。例如,Fe/ZrO2催化剂中,强金属与载体相互作用促进了缺陷氧化铁和碳化铁的生成,增强了H2/CO2的吸附活化及CO中间体解离,显著提升了低碳烯烃的产率。碳基载体则展现了不同的优势。单壁碳纳米管(SWNTs)凭借高机械强度、功能化表面和导热性,加速了CO的解离和碳化铁的形成。多壁碳纳米管(MWNTs)通过增强Fe与C的键合和表面碳氢比,抑制了CH4的生成并促进了烯烃的产率。蜂窝状石墨烯(HSG)的分级孔结构提升了反应物的渗透和活性位点的暴露,其负载的Fe催化剂展现了高活性。与传统氧化物相比,碳材料载体的可调结构为CO2加氢制烯烃提供了新策略。尽管已有显著进展,但金属与载体的协同作用机制仍需深入探索,以进一步优化催化剂设计,提升催化性能。

图10 不同载体用于提升Fe基催化剂的一些案例
新材料
碳–铁催化剂
金属氧化物负载的铁催化剂在二氧化碳直接加氢制烯烃中活性较低,归因于强金属载体相互作用。相比之下,碳基材料提供适中相互作用、还原环境高稳定性和强耐水性,促进活性相生成(图11)。研究表明,不同碳基载体对催化性能有显著影响。例如,与无定形碳壳相比,具有石墨化碳壳的催化剂减少了高级烃的形成。此外,氮掺杂碳基载体通过优化金属载体反应物相互作用,有效还原负载的铁氧化物颗粒。然而,某些氮掺杂碳材料如氮掺杂多壁碳纳米管不适合催化生产轻质烯烃,因其易生成甲烷等副产物。有序介孔碳负载的催化剂在二氧化碳加氢制液态碳方面表现优异,得益于其孔隙结构便于产物渗透。封装在氮掺杂有序介孔碳中的Fe2O3-FeCx催化剂展现了卓越的活性和稳定性。铁基催化剂的Fe3O4-FeCx异质结协同作用提升了二氧化碳制烯烃的选择性和活性。钠促进的CoFe2O4/CNT催化剂也表现出优异的二氧化碳转化率和轻质烯烃选择性,归因于双金属合金碳化物的形成。
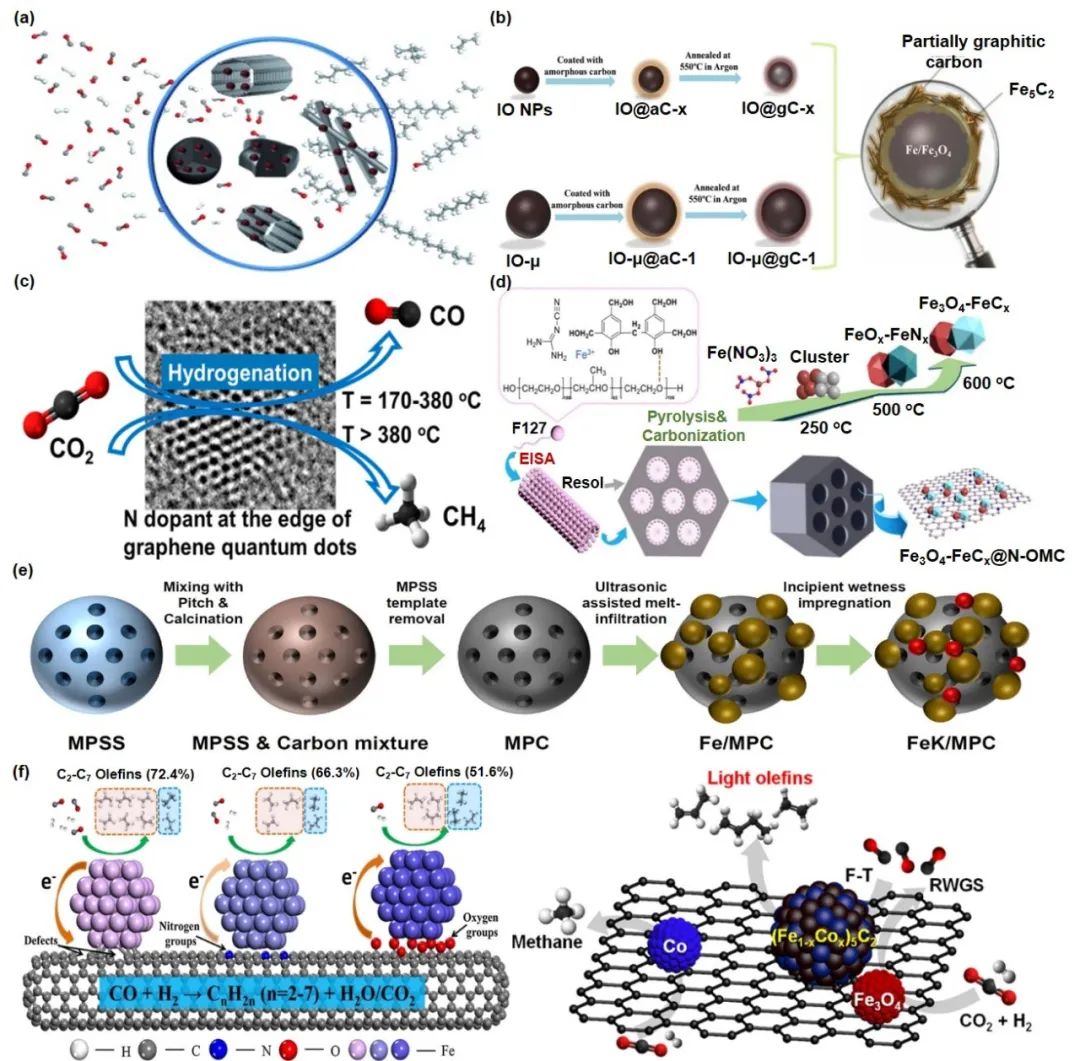
图11 碳–铁催材料用于提升Fe基催化剂的一些案例
MOF-铁催化剂
MOFs衍生多孔碳纳米材料在CO2加氢反应中展现出巨大潜力,其性能优于传统碳材料(图12)。MOFs由金属离子和有机配体构成,具有结构可调、高比表面积和明确金属位点等优势,适合作为催化载体。但其水热稳定性差,因此常通过可控热解转化为碳材料,同时嵌入金属/氧化物。与常规碳材料相比,MOFs衍生物富含氮元素,能优化催化剂表面和电子特性,提升CO2吸附和催化活性。热解过程中金属颗粒被碳层包裹,减少烧结,增强稳定性。研究表明,不同MOFs前驱体结构对催化性能有重要影响。通过调整热解温度,可调控金属纳米颗粒尺寸和孔结构。此外,引入K等掺杂剂能进一步提升烯烃选择性和催化稳定性。
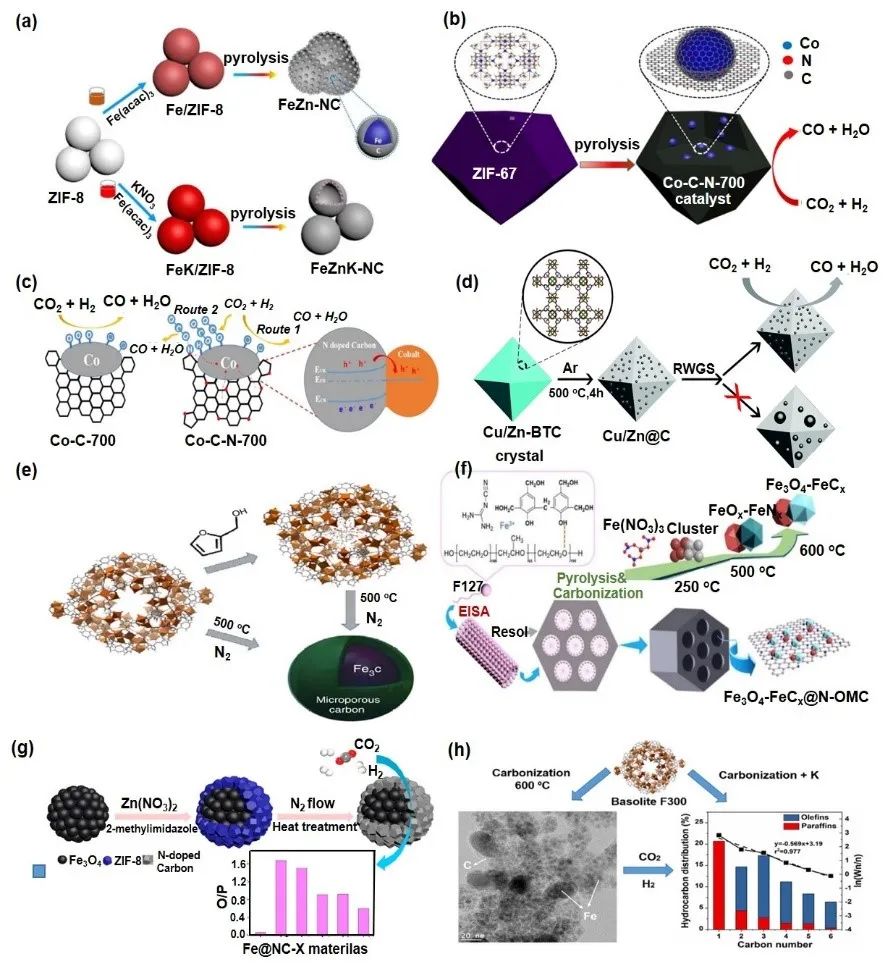
图12 MOF-铁催材料用于提升Fe基催化剂的一些案例
核壳基–铁催化剂
核壳材料在异相反应中受关注(图13)。Lin等开发的高活性F-T催化剂无需助催化剂,通过热解含铁MOF制备Fe3O4@Fe5C2核壳纳米颗粒,解决SMSI稳定颗粒但降低活性的矛盾。催化剂结构促进RWGS和FTO反应在核壳交界进行,核中RWGS生成的CO在壳层加氢为烃。FeMn@HZSM-5胶囊催化剂提高轻质烯烃选择性,降低CO2生成,HZSM-5壳抑制WGS反应。Zhang等的Fe2O3-FeCx@N-OMC催化剂,活性相封装在N-OMC中,碳化形成多种相,其中Fe3O4-FeCx异质结结构在RWGS-FTO途径中表现优异,Fe3O4催化RWGS生成CO,FeCx促进FTO反应,协同提高活性和选择性,封装结构提升稳定性。
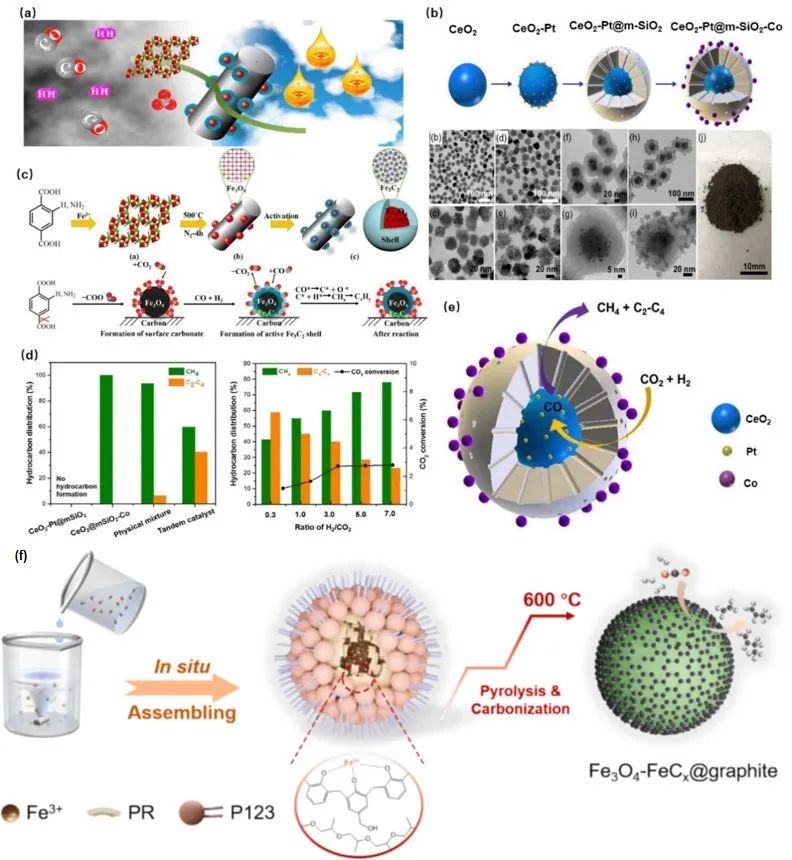
图13 核壳基–铁催化材料用于提升Fe基催化剂的一些案例
钙钛矿–铁催化剂
钙钛矿氧化物(ABO3结构)因其高热稳定性、可调物理化学性质和经济性,在许多工业应用中成为传统催化剂的可行替代品(图14)。其显著优势在于可通过全部或部分替换A和B位阳离子(包括等价或不等价替换),在保持结构完整性的同时调控稳定性、电子特性、氧化还原性和表面特性,为新型功能材料的开发提供了可能。钙钛矿可容纳阳离子和阴离子空位等固体缺陷,B位过渡金属暴露的3d电子轨道可作为活性中心。在反向水煤气变换反应中,钙钛矿衍生催化剂提供有利于CO2吸附和活化的氧空位。例如,Ma等制备的K/LaFeMnO3钙钛矿衍生催化剂在费托合成中实现了54%的轻质烯烃收率。SrFeO3作为一种钙钛矿型金属氧化物前驱体,在CO2加氢制轻质烯烃中表现突出,具有成本效益、无贵金属、高稳定性和可调化学性质。研究表明,SrFeO3可逆还原为SrO和Fe,在适当化学势下促进氧释放以分裂CO2/H2O,其还原能力和结构稳定性在宽温度范围内得以保持。此外,钙钛矿前驱体的组成和结构对Fe基催化剂的CO2加氢反应位点有深刻影响,不同价态和环境的Fe离子显著改变反应速率。Hou等报道的Sr1-xKₓFeO3钙钛矿衍生催化剂具有高度分散的活性位点和增强的反向水煤气变换活性,在CO2加氢中实现了高转化率和轻质烯烃选择性。
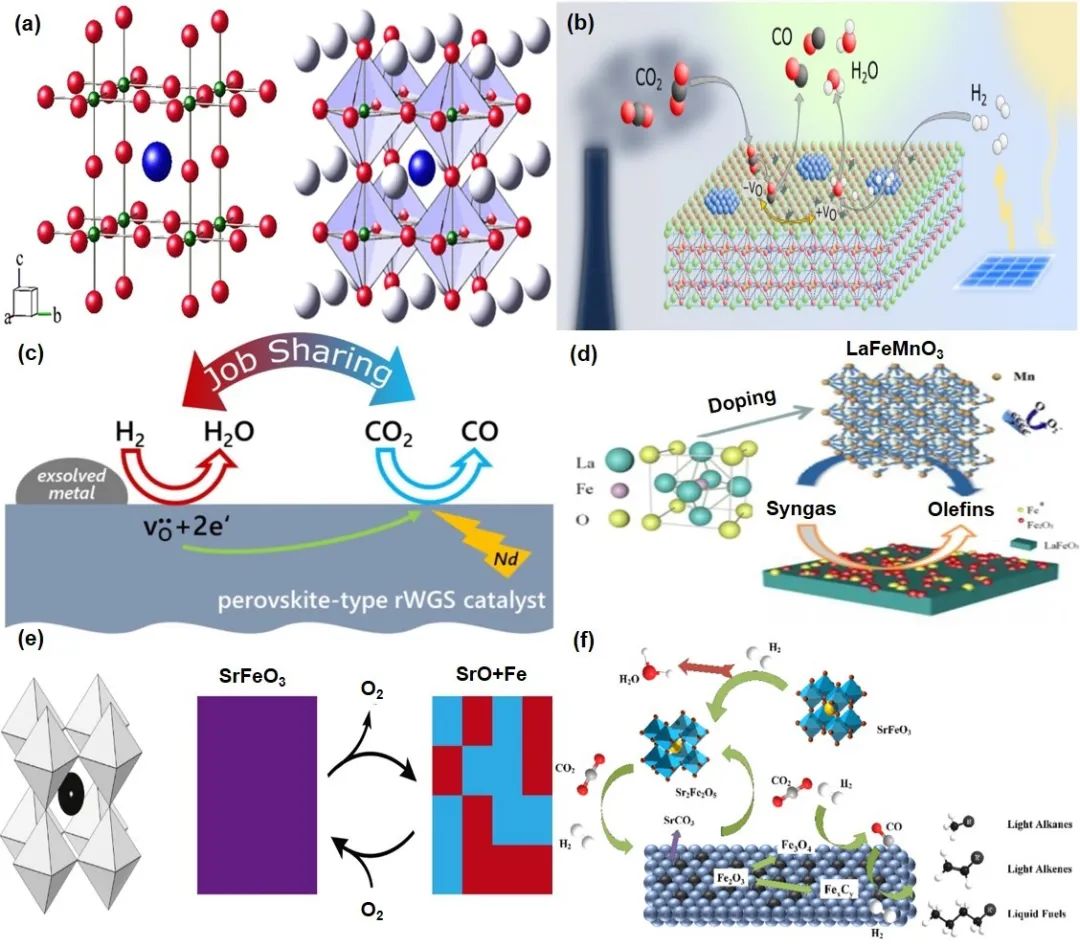
图14 钙钛矿–铁催化剂催化材料用于提升Fe基催化剂的一些案例
结论
将CO2转化为C2-C4烯烃是应对环境和工业挑战的重大进展。本文综述了CO2-MFTS路线在CO2转化中的潜力,为生产重要化工原料和减少温室气体排放提供了新途径。研究聚焦于Fe基催化剂催化CO2转化为轻烯烃的过程,分析了催化活性铁相的演变特性及其动力学和热力学行为。Fe3O4、Fe5C2等铁物种在CO2活化、加氢和烃类形成中起关键作用,但Fe5C2向Fe3O4的氧化转变是主要挑战。此外,CO2和H2分压对产物选择性和反应性的影响已通过动力学模型得到研究。碱金属、过渡金属等助剂能显著提升催化效率。尽管已有创新材料的应用,但提高C2-C4烯烃选择性、减少副产物仍需深入研究。
展望
CO2加氢制低碳烯烃前景广阔且挑战并存。未来研究方向包括:
催化剂设计与优化:稳定高效Fe基催化相,开发双功能催化剂,研究助剂影响。
催化机制深化理解:利用原位光谱等技术解析催化过程。
工艺集成与放大:整合CO2捕集与制氢技术,克服放大挑战。
经济与环境考量:降低成本,评估环境效益。
基础创新:创新催化材料与结构,提升性能与稳定性。

第一作者:刘焜
2023年武汉大学物理化学专业博士毕业,现为南昌大学资源与环境学院教师,中级职称,从事以高效、经济、环保的方式开发新型分子筛和稀土基功能材料,金属氧化物(固溶体,负载型催化剂)用于热催化CO2高效转化成高附加值化学品(烯烃、芳烃),发掘热催化二氧化碳领域中存在的关键科研问题,针对现存技术瓶颈设计、开发新型高性能催化材料。研究新型石墨材料,磷酸铁锂,磷酸铁锰材料用于锂电池正负极材料,致力于高能量密度,高稳定性性电池研究。目前以第一作者或通讯作者发表SCI论文13篇,受理公开专利4件。

通讯作者:廖光福
廖光福,博士,教授,闽江学者特聘教授,生物质基功能材料研究中心特聘教授,入选中国科协青年人才托举工程、“闽江学者奖励计划”特聘教授、福建省高层次C类人才、福建农林大学“百人攀登计划”培育项目。2020年6月于中山大学获得材料物理与化学博士学位(导师:高海洋教授);2020.年7月-2021年7月于香港中文大学机械与自动化工程系从事博士后工作(合作导师:卢怡君教授);2021年8月-2022年8月于中国地质大学担任特任研究员职位;2022年9月至今,担任福建农林大学生物质基功能材料研究中心特聘教授(陈礼辉和帅李教授团队),并于2023年4月成功入选闽江学者特聘教授,2023年12月入选中国科协青年人才托举工程。廖光福教授的主要研究方向包括光/电催化能源气体转化、聚合物合成与应用、氧化还原液流电池、纳米结构材料、气体储存和能量转换等。目前主持国家自然科学基金青年项目1项,主持福建省自然科学基金1项,至今已经在Progress in Materials Science (影响因子48.165)、Energy & Environmental Science(影响因子39.714)、 Physics Reports(影响因子30.510)、Matter、Chemical Science、ACS Catalysis、Nano Energy、Macromolecules、Small、Journal of Materials Chemistry A、Journal of Catalysis、Chemical Engineering Journal、Nano Reseach等国际著名刊物发表SCI论文60余篇,其中高被引论文7篇,授权专利10件。论文总引用3473次,H-index 30。并长期担任ACS Nano、Macromolecules、Applied Catalysis B: Environmental、ACS Applied Materials & Interfaces、Chemical Engineering Journal、The Journal of Physical Chemistry C等国际著名期刊审稿人,每年审稿约50~60篇。并担任Advanced Fiber Materials、eScience、Exploration青年编委,也成功入选了2022年Journal of Materials Chemistry A新锐科学家和2023年Chemical Communications新锐科学家。课题组网站如下所示:http://www.polymer.cn/ss/liaogf/index.html.
Kun Liu*, Muhammad Asif Nawaz, Guangfu Liao*. Decoding fundamental insights and outlooks on state-of-the-art iron-catalyst design strategies for meliorated CO2 valorization into light olefins. Coordination Chemistry Reviews, 2025, 216611.DOI: 10.1016/j.ccr.2025.216611