

电荷密度差分(Charge Density Difference)是分析材料界面电荷转移、吸附体系电子重分布的重要手段。本文将手把手教你用VASP完成电荷密度差分计算,涵盖参数设置、数据处理及可视化全流程,确保每一步均可复现!


一、什么是电荷密度差分?
电荷密度差分描述体系中电子密度的重新分布情况,计算公式为:
Δρ = ρ(AB) – ρ(A) – ρ(B)
其中:
· ρ(AB):复合体系(如异质结/吸附体系)的总电荷密度
· ρ(A)和ρ(B):孤立组分A和B的电荷密度
典型应用场景:
单原子催化剂表面吸附、异质结界面电荷转移、掺杂材料电子结构分析等等。
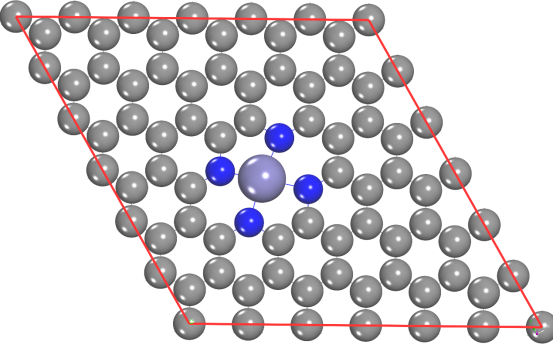
二、计算步骤详解
步骤1:结构优化(关键!)
目标:获得所有体系的稳定几何结构
操作流程:
1.优化复合体系AB
o 构建AB模型(如吸附结构、异质结)
o INCAR关键参数:
IBRION =2 # 使用准牛顿算法优化
NSW =100 # 最大离子步数
EDIFFG =-0.02 # 收敛标准(力2 eV/Å)
ISIF =2 # 优化原子位置
2.不要优化孤立组分A和B
o 保持与AB相同的计算参数(ENCUT、K点等)只做单点能计算
注意:必须保证体系A和体系B的原子位置与AB完全一致!
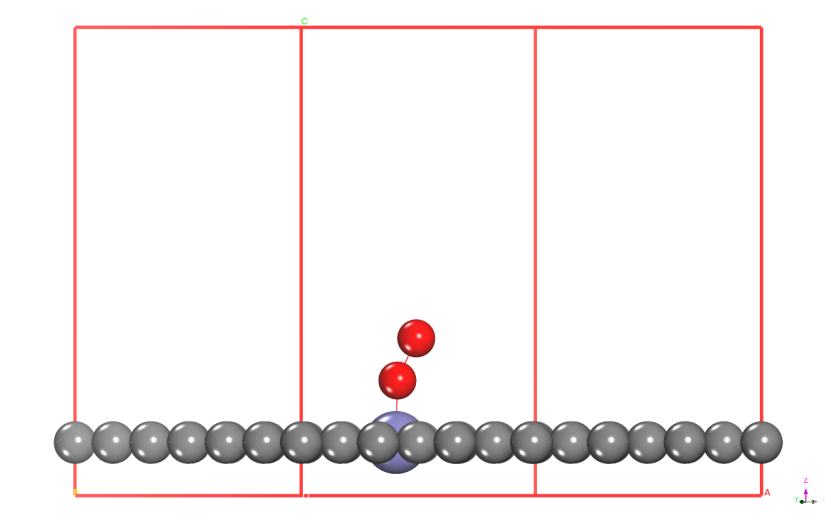
步骤2:静态自洽计算
目标:获取高精度电荷密度文件CHGCAR
操作流程:
1.对AB、A、B分别进行静态计算
o 使用优化后的结构
o INCAR关键参数:
ISTART=1 # 读取波函数加速收敛(若从WAVECAR重启)
ICHARG = 1
NSW =0 # 关闭离子弛豫
LCHARG = .TRUE. # 必须输出CHGCAR
PREC = Accurate # 高精度模式
NGXF = 320
NGYF = 320
NGZF = 320 #确保每个计算中网格密度参数设置一致
2.统一参数设置
o 确保所有体系的ENCUT、网格密度、真空层厚度完全一致
o 建议使用Gamma中心K点生成方式

步骤3:电荷密度差分计算
核心操作:使用CHGCAR文件生成差分电荷密度
操作步骤:
1.文件准备
o 将AB、A、B的CHGCAR分别重命名为:
CHGCAR_AB
CHGCAR_A
CHGCAR_B
2.执行差分计算
o 运行chgsum.pl脚本获得CHGCAR_sum:
Chgsum.pl CHGCAR_A CHGCAR_B
3.执行差分计算
o 运行chgdiff.pl脚本计算电荷密度差分:
Chgdiff.pl CHGCAR_sum CHGCAR_AB
4.输出结果
o 生成CHGDIFF.diff(差分电荷密度文件)
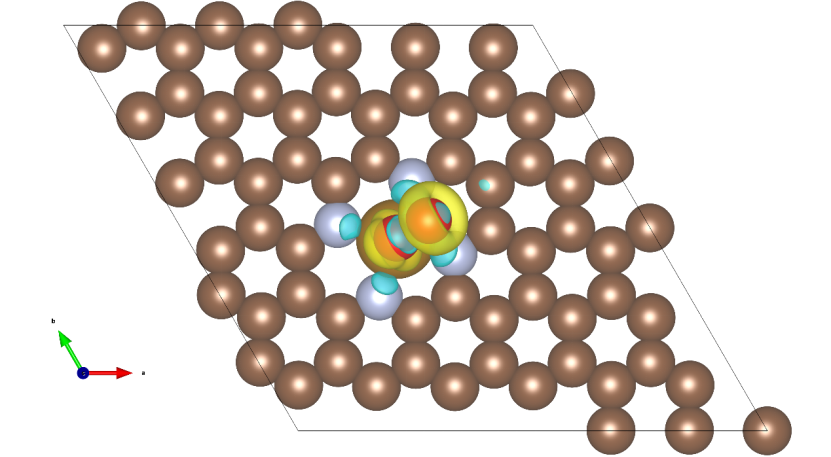
步骤4:可视化分析
推荐工具:VESTA(免费开源)
操作流程:
1.用VESTA打开CHGDIFF.diff
2.设置等值面:
o 点击Properties → Isosurfaces
o 正值为电子积累(建议设等值面0.0015 e/ų,黄色)
o 负值为电子流失(建议设等值面-0.0015 e/ų,蓝绿色)
3.调整显示效果:
o 使用Properties→ Radius and color修改原子颜色和大小
o 通过Edit→ Lattice Planes添加切片视图
三、常见问题FAQ
1.为何要保证K点网格密度一致?
o 网格密度不同会导致电荷密度网格不匹配,无法直接相减。
2.差分电荷密度数值范围如何选择?
o 一般取±0.005 e/ų,具体可根据体系调整。
四、注意事项
· 结构优化不充分会导致错误结果,务必检查OSZICAR中的收敛标志。
· 真空层厚度不足会导致镜像电荷干扰,建议真空层≥15 Å。
找华算做计算👍专业靠谱省心又省时!
益于理论计算化学的快速发展,计算模拟在纳米材料研究中的运用日益广泛而深入。科研领域已经逐步形成了“精准制备-理论模拟-先进表征”的研究模式,而正是这种实验和计算模拟的联合佐证,更加增添了论文的可靠性和严谨性,往往能够得到更广泛的认可。
华算科技已向国内外1000多家高校/科研单位提供了超过50000项理论计算和测试表征服务,部分计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。
