

作为实验研究人员,可能大家对理论计算的具体应用和选择还不是很清楚。因此需要仔细思考,哪些理论计算能够有效支持实验结果,并提升论文的说服力。比如说研究主要集中在开发一种新型的非贵金属电催化剂,用于氧还原反应(ORR)。通过实验,已经获得了催化剂的活性数据(如半波电位、塔菲尔斜率)、稳定性测试结果以及通过XPS、TEM等表征手段得到的材料结构信息。现在,需要理论计算来解释这些实验现象背后的机理,并验证催化剂的活性位点和反应路径。
那么,哪些理论计算方法适用于实验研究的数据适配呢?
1. 活性位点识别与结构优化
计算内容:
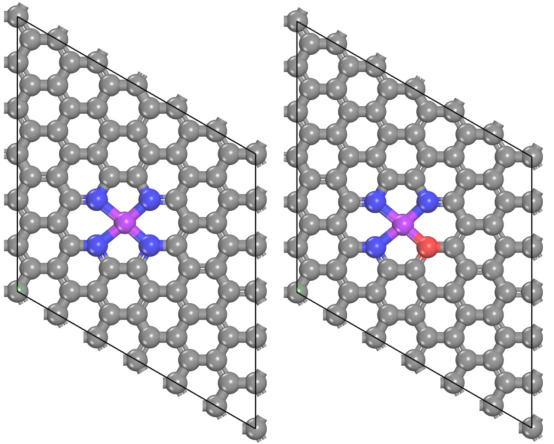
· DFT结构优化:构建催化剂的原子模型(如Fe-N₄、Fe-N₃O等),优化几何结构至能量最低态。
· 形成能计算:比较不同配位结构的形成能,确定热力学最稳定的活性位点。

适配实验数据:
· XPS/XAS表征:通过计算Fe的氧化态(如Fe²⁺或Fe³⁺)与配位环境,验证实验中的金属价态和配位结构。
· EXAFS拟合:对比理论模型中的键长(如Fe-N键长)与实验拟合结果,确认活性位点的原子排列。
选择原因:
明确活性位点的几何和电子结构是解释催化活性的基础,直接关联实验表征中的化学状态信息。
2. 反应路径与自由能分析
计算内容:
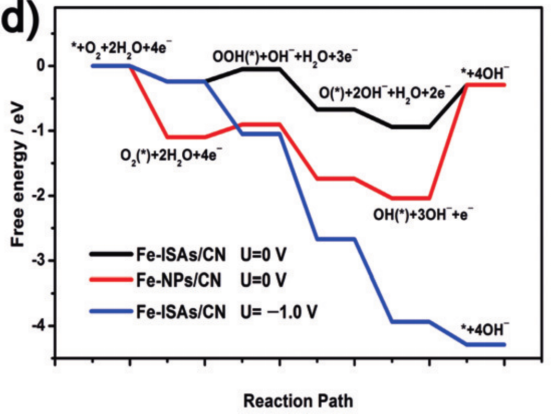
· 自由能台阶图:计算ORR各步骤(O₂吸附→*OOH→*O→*OH→OH⁻)的吉布斯自由能变化(ΔG)。
· 决速步骤(RDS)识别:通过能垒最高的步骤确定反应动力学瓶颈。
适配实验数据:
· Tafel斜率:理论计算的RDS与实验Tafel斜率对应的反应机制(Volmer、Heyrovsky或Tafel步骤)需一致。
· 过电位(η):通过理论过电位(η = max(ΔG/e) – 1.23 V)验证实验测量的极化曲线。
选择原因:
自由能分析定量揭示反应路径的可行性,解释实验活性差异(如不同催化剂的半波电位差异)。
3. 电子结构计算
计算内容:
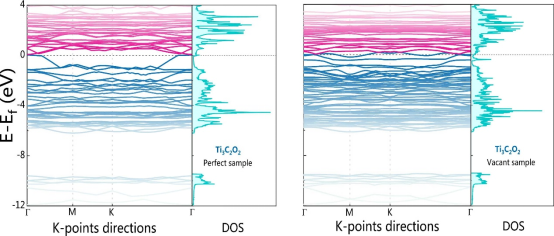
· 态密度(DOS)与能带结构:分析催化剂费米能级附近的电子态分布,评估导电性。
· 电荷密度差与Bader电荷:研究活性位点与反应中间体(如*O、*OH)的电荷转移。
适配实验数据:
· 电化学阻抗谱(EIS):高导电性催化剂的理论DOS应显示金属性或窄带隙,与低电荷转移电阻(Rct)的实验结果对应。
· XPS价带谱:计算DOS与实验价带谱的匹配验证电子结构模型的准确性。
选择原因:
电子结构决定催化剂的电荷传输能力,直接影响反应动力学。
4. 吸附能与中间体稳定性
计算内容:
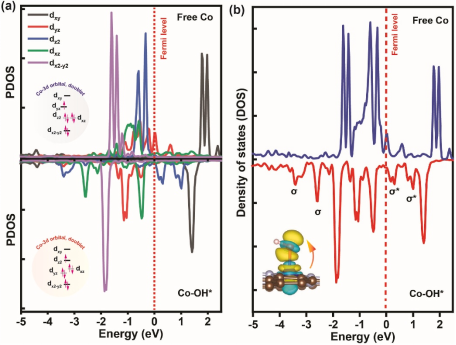
· 中间体吸附能计算:计算O₂、*OOH、*O、*OH在活性位点的吸附能(E_ads)。
· d带中心理论:分析过渡金属(如Fe)的d带中心位置,预测吸附强度。
适配实验数据:
· 活性-选择性关系:弱*OH吸附(ΔG_OH接近0 eV)对应高ORR选择性和低过电位,与实验的旋转圆盘电极(RDE)测试结果关联。
· 原位红外/拉曼光谱:理论预测的中间体(如*OOH)应与实验观测到的振动峰匹配。
选择原因:
吸附能是催化活性的核心描述符,直接关联Sabatier原理(最佳吸附强度)。
5. 稳定性与降解机制分析
计算内容:
· 溶解电位计算:通过Pourbaix图预测催化剂在特定pH和电位下的稳定性。
· 氧空位形成能:评估催化剂在氧化环境中的结构退化倾向。
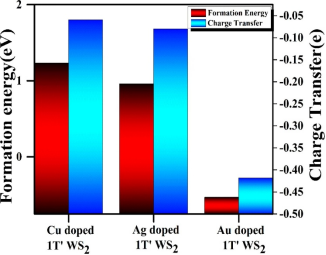
适配实验数据:
· 加速耐久性测试(ADT):高溶解电位或低氧空位形成能对应实验中的稳定性提升(如5000圈CV后活性衰减
· TEM/STEM表征:计算模拟的结构降解路径(如Fe浸出)需与实验观察的形貌变化一致。
选择原因:
稳定性计算为实验耐久性提供理论预测,指导材料设计。
6. 溶剂化与电场效应修正
计算内容:
· 隐式溶剂模型(如VASP中的VASPsol):修正吸附能以考虑电解液的屏蔽效应。
· 外加电场模拟:研究电极电位对反应能垒的影响(如通过计算氢电极模型,CHE)。
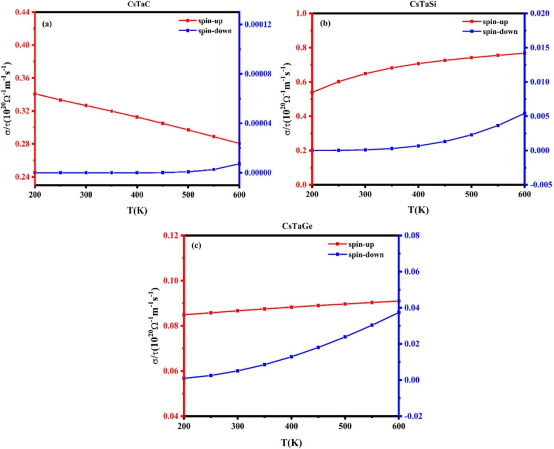
适配实验数据:
· pH依赖性活性:理论计算的电位-pH关系应与实验极化曲线的pH依赖性一致。
· 双电层效应:修正后的吸附能更贴近实际电化学界面环境。
选择原因:
溶剂化和电场效应是电催化区别于热催化的核心,提升计算结果的实验相关性。
7. 机器学习辅助高通量筛选(可选)
计算内容:
· 描述符数据库构建:基于DFT计算生成吸附能、d带中心等关键参数数据库。
· 机器学习模型训练:预测新型催化剂的活性与稳定性。
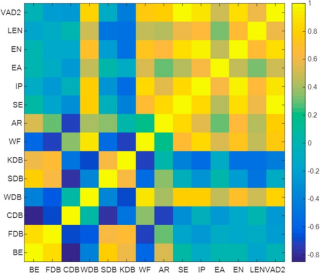
适配实验数据:
· 催化剂组分优化:指导实验合成(如预测Fe-Co双金属位点的协同效应)。
选择原因:
加速材料发现,为后续实验提供理论指导。
总结:理论计算与实验的协同作用
· 机理验证:理论计算从原子尺度揭示实验现象的本质(如活性位点、反应路径)。
· 性能预测:通过计算描述符(吸附能、d带中心)指导催化剂设计,减少实验试错成本。
· 数据互补:实验表征(XPS、TEM)与理论模型(电荷分布、结构优化)相互印证,提升论文可信度。
通过上述计算内容,可系统构建“结构-性能-机理”的理论框架,为实验数据提供坚实的理论支撑,显著提升论文的科学价值。
找华算做计算👍专业靠谱省心又省时!
益于理论计算化学的快速发展,计算模拟在纳米材料研究中的运用日益广泛而深入。科研领域已经逐步形成了“精准制备-理论模拟-先进表征”的研究模式,而正是这种实验和计算模拟的联合佐证,更加增添了论文的可靠性和严谨性,往往能够得到更广泛的认可。
华算科技已向国内外1000多家高校/科研单位提供了超过50000项理论计算和测试表征服务,部分计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。
