

引言:电催化析氧反应(OER)为何如此重要?
在可再生能源技术(如电解水制氢、金属–空气电池)中,析氧反应(OER)是关键的阳极半反应。然而,OER涉及四电子转移过程(4OH⁻ → O₂ + 2H₂O + 4e⁻),动力学缓慢且过电势高,成为制约能量转换效率的瓶颈。开发高效、稳定的电催化剂是解决这一问题的核心,而密度泛函理论(DFT)计算已成为揭示催化机理、指导实验设计的“理论显微镜”。
第一部分:DFT计算在OER研究中的核心作用

1.1 理论框架:从薛定谔方程到Kohn-Sham方程
DFT基于量子力学第一性原理,通过求解Kohn-Sham方程,将多电子体系的基态能量表示为电子密度的泛函。对OER研究而言,关键参数包括:
·*吸附能(ΔG)**:中间物种(*O、*OH、*OOH)在催化剂表面的吸附自由能,决定反应路径和过电势。
· d带中心理论:过渡金属催化剂的d带中心位置影响中间体吸附强度,调控活性(Sabatier原理)。
· 电荷转移与轨道杂化:催化剂表面与中间体的电子相互作用(如Co³⁺的eg轨道占据数)。
1.2 计算挑战与解决方案
·溶剂化效应:隐式溶剂模型(如VASPsol)或显式水分子模型需引入以模拟真实电解液环境。
·电势依赖性:通过计算氢电极(CHE)模型或显式电荷法(如恒电势法)关联理论过电势与实验值。
·自旋极化与磁态:过渡金属氧化物(如Fe/Ni基催化剂)需考虑自旋态对活性的影响。
第二部分:OER反应机理的DFT解析
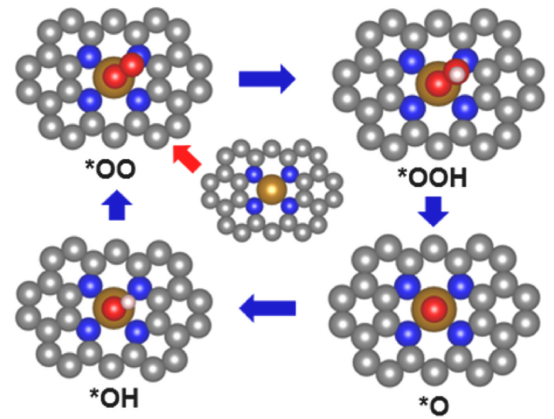
2.1 经典四步反应路径
以金属氧化物(如IrO₂、RuO₂)为例,OER通常遵循以下步骤:
1.H₂O解离吸附:H₂O → *OH + H⁺ + e⁻
2.*OH脱质子:*OH → *O + H⁺ + e⁻
3.O-O键形成:*O + H₂O → *OOH + H⁺ + e⁻
4.O₂脱附:*OOH → O₂ + H⁺ + e⁻
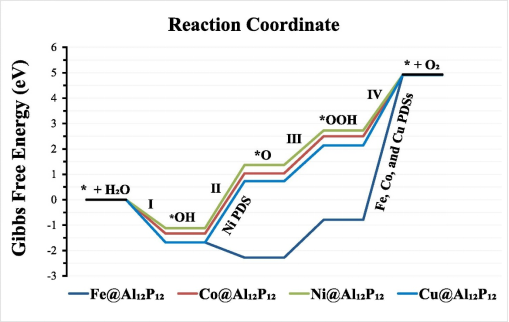
**决速步(RDS)**通常为第三步(*O → *OOH),其自由能变化(ΔG3)决定理论过电势(η = max{ΔG1, ΔG2, ΔG3, ΔG4}/e – 1.23 V)。
2.2 替代反应路径的竞争
· 晶格氧参与机制(LOM):某些钙钛矿(如SrCoO₃)中,晶格氧直接参与O-O键形成,可能绕过*OOH生成步骤,降低过电势(需通过氧空位形成能验证)。
· 双位点协同催化:如NiFe-LDH中,Ni位点吸附O,Fe位点促进OOH形成。
第三部分:催化剂理性设计的DFT指导策略
3.1 电子结构调控
· 掺杂效应:引入杂原子(如Mo掺杂Co3O4)可优化d带中心,削弱*O吸附(案例:Nature Catalysis, 2021)。
· 应变工程:拉伸应变使NiOOH的Ni-O键长增加,降低ΔG3(Phys. Rev. B, 2020)。
3.2 表面缺陷工程
· 氧空位(Vo):增强电荷转移,促进H₂O解离(如CeO₂-Vo体系,JACS 2022)。
· 台阶边缘与晶界:高指数晶面暴露更多活性位点(如PtNi@NiO核壳结构)。
3.3 多尺度模拟结合
· 高通量筛选:基于Materials Project数据库,结合描述符(如ΔGO – ΔGOH)筛选潜在催化剂。
· 机器学习加速:利用神经网络势函数(如DeepMD)实现长时程动力学模拟,预测稳定性。
第四部分:前沿进展与挑战
4.1 动态过程与真实条件模拟
· 电势–界面耦合:采用Poisson-Boltzmann方程修正表面电荷分布。
· 温度与压力效应:过渡态理论(TST)结合分子动力学(AIMD)模拟实际工况。
4.2 非晶态与单原子催化剂
· 非晶Co-B催化剂:短程有序结构提供多样化活性位点。
· 单原子催化剂(SACs):如Fe-N-C中单原子Fe的配位环境调控。
结语:理论与实验的协同创新
DFT计算不仅为理解OER机理提供原子级视角,更通过“计算先行–实验验证”的闭环加速新材料开发。随着算力提升与算法革新(如量子嵌入理论、多物理场耦合),理论电化学正迈向更高精度与更广应用边界。
互动话题:你认为未来OER催化剂设计的突破口在哪里?欢迎在评论区留言讨论!
益于理论计算化学的快速发展,计算模拟在纳米材料研究中的运用日益广泛而深入。科研领域已经逐步形成了“精准制备-理论模拟-先进表征”的研究模式,而正是这种实验和计算模拟的联合佐证,更加增添了论文的可靠性和严谨性,往往能够得到更广泛的认可。
华算科技已向国内外1000多家高校/科研单位提供了超过50000项理论计算和测试表征服务,部分计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。
