



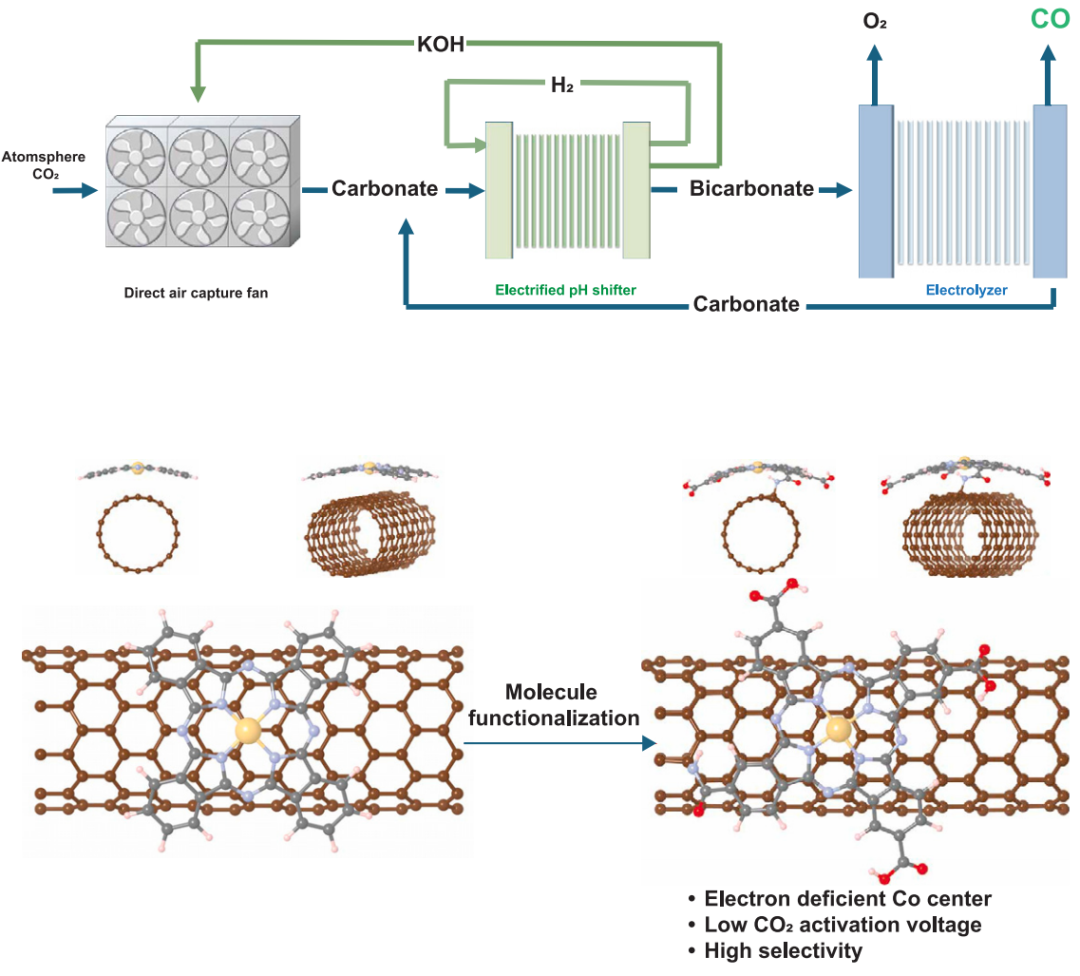
电气化反应捕集可将从空气捕集后的碳酸碱液体中的二氧化碳升级为增值产品,同时再生捕集介质。由于高的电解电压(>3.7 V)和适中的CO选择性(
美国西北大学Edward H. Sargent院士、Xie Ke等人开发了一种Co分子催化剂,具有缺电子的Co中心,降低了所需的还原电压。然后将催化剂接枝到导电载体上,增强电荷转移。电气化pH降低装置提高了CO2的利用率,提高了CO的选择性。该系统在2.7 V和100 mA cm-2下实现了70%的CO选择性,对应的能量强度为35 GJ/t CO。该技术的能源成本与直接空气捕集(DAC)然后进行逆水气转换(RWGS)相当,但它可在室温操作。
相关工作以《Electrosynthesis of CO from an electrically pH-shifted DAC post-capture liquid using a catalyst:support amide linkage》为题在《Joule》上发表论文。值得注意的是, 这也是Edward H. Sargent院士在《Joule》上发表的22篇论文。
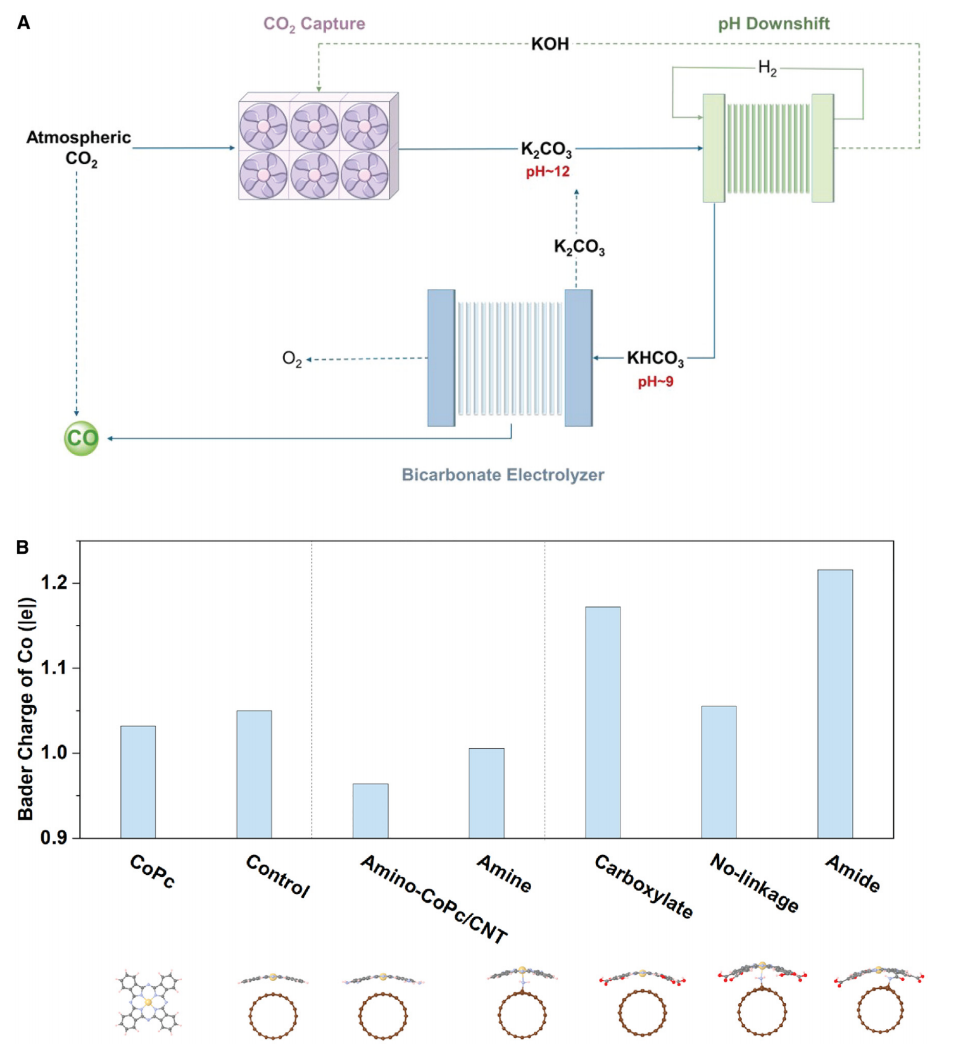
图1 通过添加不同官能团调制CoPc的电子结构
本文探索了一种全电气化系统的使用(图1A),该系统包括第一阶段,将pH值降低到碳酸氢盐的pH值,然后将这种液体中的CO2还原为CO。目前最节能的碳酸氢盐电解槽的全电解电压通常在3.2~3.8 V范围内,电流密度为100 mAcm-2。因此,在此将重点放在降低工作电压上。
二氧化碳活化的阴极过电位是电解电压的主要影响因素,可以通过电子态调节来降低;在这里,使用酞菁钴(CoPc),通过引入官能团来操纵其电子性质的分子结构。研究了用供电子基团(记为“amino-CoPc/CNT”)和吸电子基团(记为“carboxylate”)功能化CoPc,旨在调节钴中心的电子状态。将这些功能化分子沉积在碳纳米管上,然后还研究了CoPc与碳纳米管载体之间的化学联系。为了初步评估官能团对CoPc的Co中心的影响,使用了DFT进行分析:图1B中Co位点上的Bader电荷表明羧酸盐诱导Co中心的电子耗尽,而胺基诱导富电子条件。
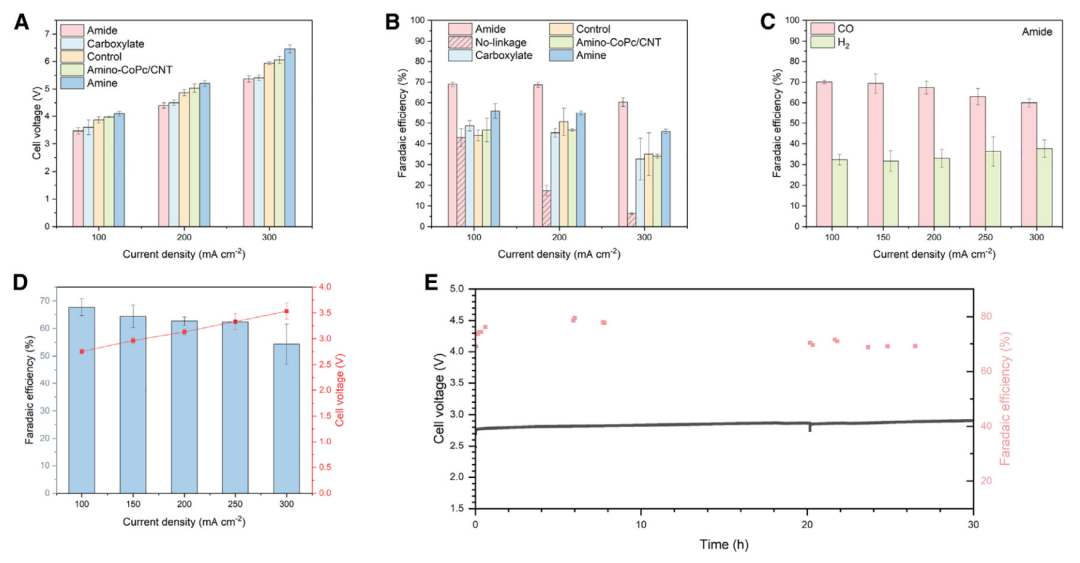
图2 碳酸氢盐电解性能与CoPc和CNTs上官能团选择的关系
使用商业双极膜(BPM)筛选了双电极膜电极组件(MEA)电解槽中碳酸氢盐的电解性能;在BPM和阴极之间夹入一层85 mm厚的聚四氟乙烯(PTFE)多孔中间层。可以观察到(图2A)羧酸盐降低过电位,而amino-CoPc/CNT对过电位几乎没有影响。与受益于CoPc分子功能化的“对照”相比,这两种功能化都不能提高CO的选择性(图2B);在100 mA cm-2时,它们都显示出相似的46%-51%的FECO。
假设,如果能在碳纳米管载体和钴中心之间实现有效的电荷转移,CO选择性可能会得到改善。碳纳米管载体上的官能团和CoPc之间的共价键,如酰胺键,可能潜在地实现这一目标。这类样品称之为“amide”(CNT-NH2上的CoPc-COOH键)。它们表现出与羧酸盐类似的低过电位(图2A)。然而,在100 mA cm-2时,amide同时显示FECO增加~70%(图2B);在CNT-NH2样品上制备了CoPc-COOH,没有形成酰胺键(表示为“no-linkage”),并记录了FECOCO的变化不显著(图2B)。这些结果突出了酰胺键的关键作用。
为了构建一个完整的电解槽,使用了一个NiFeOx电极和一个BPM,使用TiO2作为水解离催化剂,在100 mA cm-2下获得了电解电压2.7 V(图2D)。通过观察碳酸氢盐电解槽在100 mA cm-2电流密度下的连续运行,研究了电解槽的耐久性。发现运行超过30小时FECO可保持70%(图2E)。在之前的一些关于电化学反应捕集的报告中,有富含碳酸盐的捕集后液体被送入基于BPM的电解槽,在那里碳酸盐被转化为二氧化碳并还原为产品。在这里,在前端添加了一个子系统(pH-downshifter)(图1A),以在捕获后溶液~12和碳酸氢盐~9的pH值之间改变电解溶液。较低的pH值促进了i-CO2的可用性,但消耗了更多的电力。因此,用不同的pH调节器出水pH值来评估每个步骤的用电量。
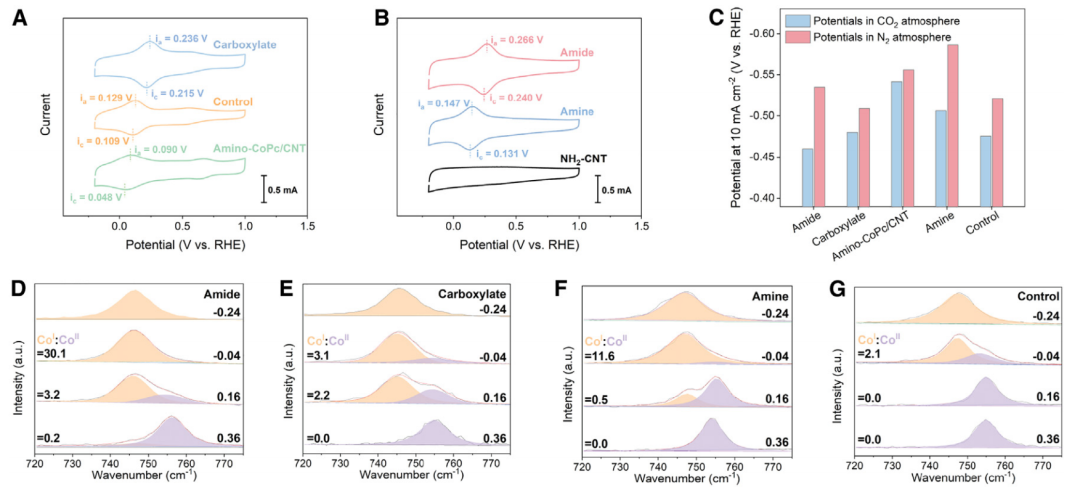
图3 功能化CoPc/CNT样品的电子结构调制
本文采用CV对其机理进行了研究。比较了CoPc与不含官能团的情况,发现在Ar饱和电解质中,所有样品都表现出0.048至0.266 V之间的可逆氧化还原电对,分配给CoII/CoI氧化还原。CoII需要转化为CoI才能对CO2还原有活性。在这些样品中,羧酸盐的CoII/CoI氧化还原峰与对照相比出现了正位移(图3A),这意味着Pc配体上的-COOH基团将电子从钴中心抽离。相比之下,amino-CoPc/CNT的CoII/CoI氧化还原与对照以及羧酸盐相比呈现负位移。这一结果与它们相似的电压趋势(图2A)是一致的,吸电子特性与这些材料中的过电位降低有关。
通过CO2和N2气氛下的LSV测试(图3C)比较了CO2R活性。这与MEA中电解电压的趋势非常吻合(图2A)。在CO2中,amide显示出较低过电位。在N2气氛下,amide表现出比CO2气氛下更高的析氢反应电位,表明其CO2还原动力学比HER更快。相比之下,carboxylate在CO2和N2气氛中表现出相似的电位。这些结果与观察到的酰胺较高的FECO一致。
使用原位拉曼光谱(图3D-3G)来探测CoII(拉曼位移:754 cm-1)到CoI(拉曼位移:747 cm-1)的转变。对于amide,carboxylate和amine,CoII在0.16 V时开始转化为CoI。在0.16 V时,amide的CoI:CoII较高为~3.2,carboxylate为2.2,amine为0.5。在0.04 V条件下,与carboxylate(3.1)和amine(11.6)相比,amide的CoI:CoII比为30,amide的CoII转化为CoI更完全。在-0.24 V下,所有样品中的CoII均不存在。拉曼结果与CV数据一致,表明CoII向CoI转化的趋势遵循难度增加的顺序:amide(最容易)
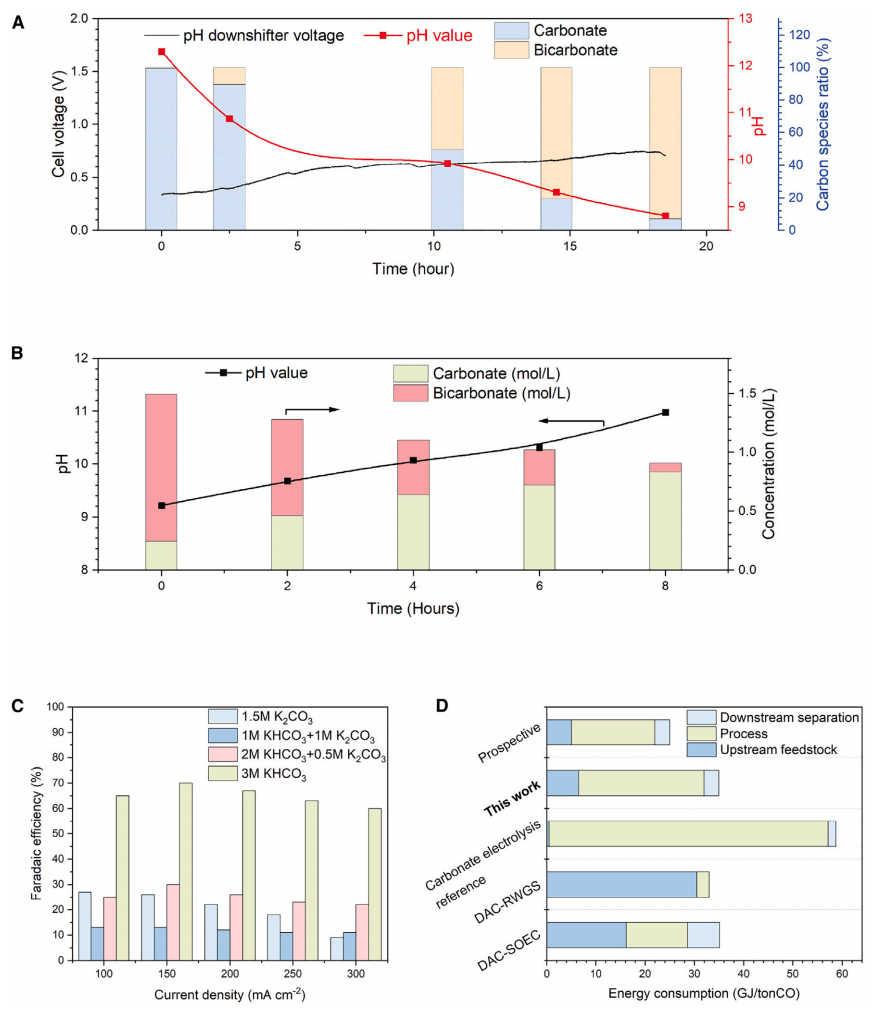
图4 系统演示和能量评估
使用由1.5 M K2CO3溶液组成的模拟捕获后液体注入pH调节系统,并监测阳极电解质pH(以及碳酸盐:碳酸氢盐的相对组成)以及电解电压(图4A)。在最终pH值为~9的情况下,该装置将93%的碳酸盐转化为碳酸氢盐,并且在此过程中消耗3.8 GJ/吨CO2(6 GJ/吨CO)的电力。同时,KOH在阴极电解质中再生,接近生成的碳酸氢盐的化学计量。
使用pH为~9的处理后溶液作为原料进行连续碳酸氢盐电解。pH值的变化和相应的碳酸氢盐浓度随时间的变化如图4B所示。由此得出结论,溶解的无机碳转化为CO,而碳酸盐在系统的液相出口被输送。电解后的溶液再次输入pH调节器,以恢复pH为~9,并再次演示CO的生产。
纵观整个过程,可以发现,由于碳酸氢盐电解占总能耗的主导地位,并且由于碳酸氢盐电解比碳酸氢盐电解效率高得多,因此将碳酸氢盐的pH值一直调到8.8是有意义的。在这种情况下,电解能量为25 GJ/吨CO,总能量为35 GJ/吨CO。通过热释放(钙环)产生浓气相CO2,然后进行气相CO2共电解或逆水气转换(RWGS)等替代方法,发现目前的实验数据接近最有效的竞争者DAC+RWGS系统的数据。