量子化学作为一种研究原子和分子行为的理论方法,在化学、生物学、材料科学等领域具有广泛的应用。
通过使用量子化学,我们可以从理论层面解释实验现象、理解化学反应机理、预测分子性质、设计新型催化剂和药物、探索材料的电子结构和性能。
Gaussian 16是用于计算分子和材料性质的量子化学软件。它使用各种理论方法和近似来解决包括能量、结构、振动频率、光谱性质等在内的多种化学问题。
Gaussian 16支持包括从简单的Hatree-Fock (HF) 方法到更精确的密度泛函理论 (DFT) 和耦合簇 (CCSD(T)) 方法。
此外,通过可视化工具GaussView,可以直观分析计算结果。由于广泛的应用和灵活的功能,Gaussian程序在科学研究、药物设计、材料科学等领域都有着不可替代的作用。
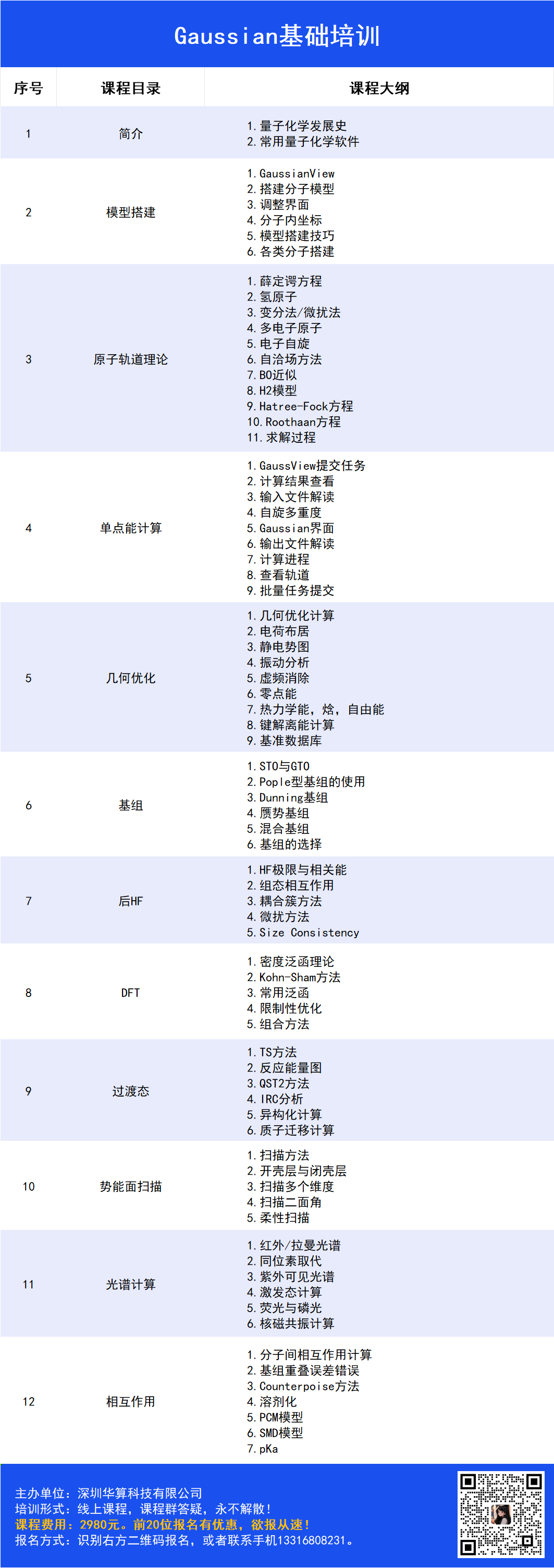
Gaussian 16与GaussView
Gaussian的涉及量子化学理论,内部公式推导繁多,而软件本身经数十年发展已包含多种方法与基组,相关的选择也常令初学者疑惑。配套可视化工具GaussView由于包含多样的建模技巧,初学者也难以快速掌握。
为了有效降低大家入门Gaussian的门槛,华算科技黄老师原创设计了Gaussian零基础课程,课程同时包含理论与实操部分,并包含大量化学性质的计算案例,旨在帮助大家快速掌握Gaussian这一有力工具,并快速使用到自己的研究之中。
前20位报名立减300元,数量有限,先到先得!
报名方式:识别下方二维码报名,或者联系手机133-1680-8231。
👇👇扫描二维码,立即报名👇👇
👆👆电话:133-1680-8231👆👆
黄老师:华算科技全职技术专家,武汉大学本科,北京大学博士,新加坡国立大学访问学者。目前已发表SCI文章共20篇,其中第一作者文章5篇,单篇最高影响因子>40。
从事理论计算与实验化学研究工作十二年,擅长使用Gaussian进行化学体系的研究,数据处理,机器学习等。
入门到精通!Python/机器学习系列课程:Python数据分析、机器学习与材料/化学、神经网络、机器学习与电催化等!
小白友好!快速入门计算!MS+LAMMPS建模/催化/电池/半导体计算培训…零基础到进阶!
彻底搞定DFT计算!VASP计算十四大专题课程:金属、晶体、二维材料、催化、电池、钙钛矿、单原子、吸附、磁性、半导体缺陷计算等!
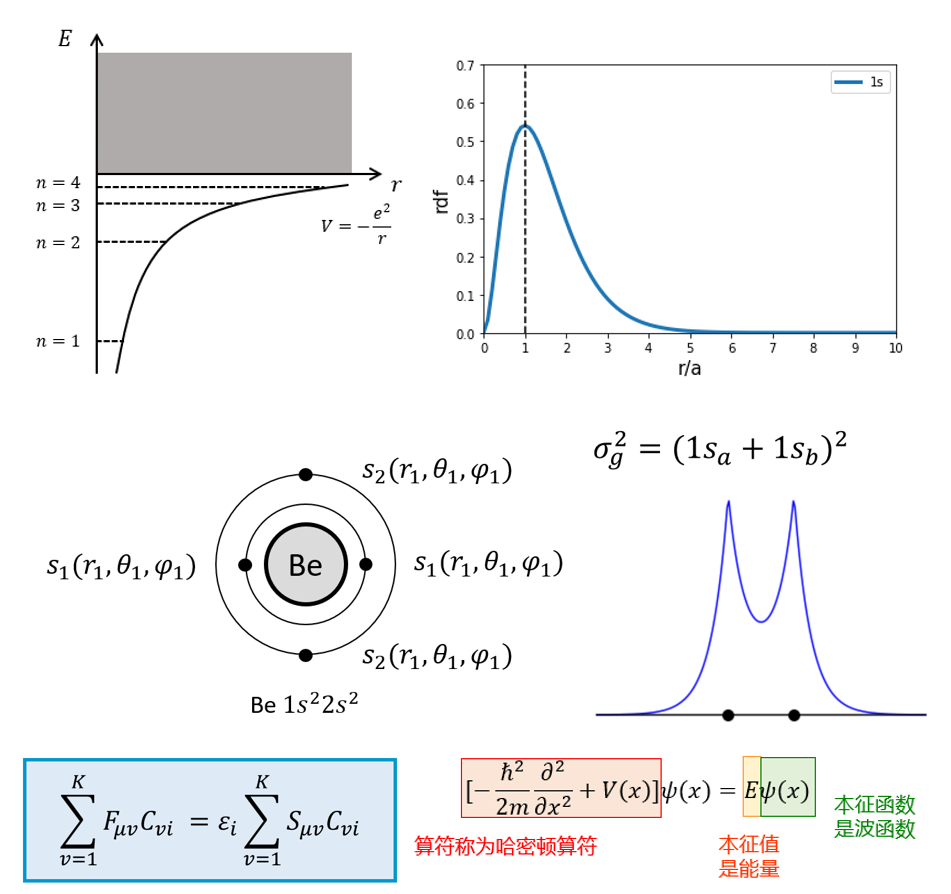
快速了解薛定谔方程、Hatree-Fock方程、Roothaan方程的来源与求解方法。
学习Hatree-Fock方程的解析解法与数值解法。
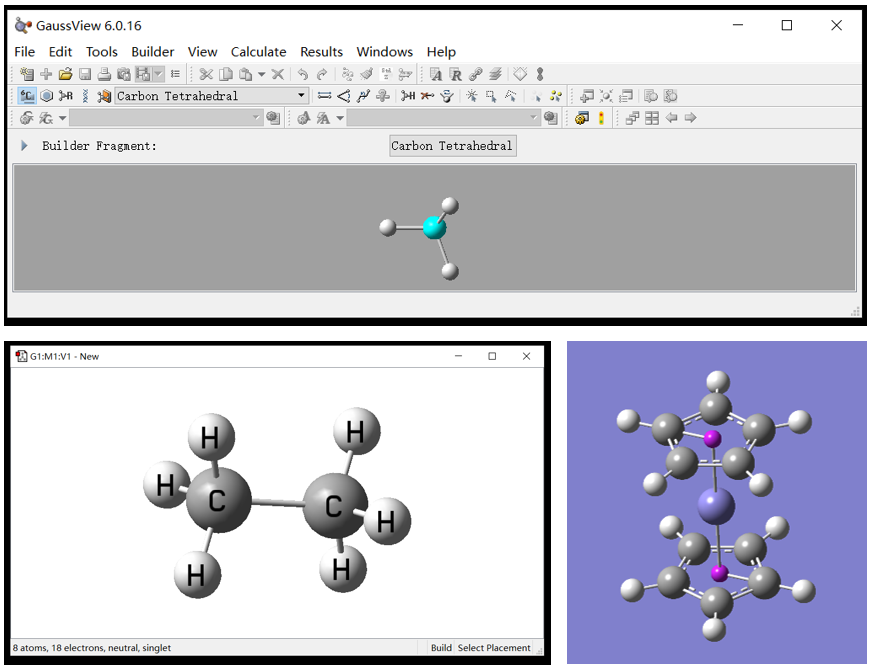
GaussView常用于快速构建分子结构,是分子建模的利器。
本节将学习GaussView的基本使用,分子基本查看、调整方法。
通过实操介绍常规模型构建方法,各类模型的显式与结构保存。
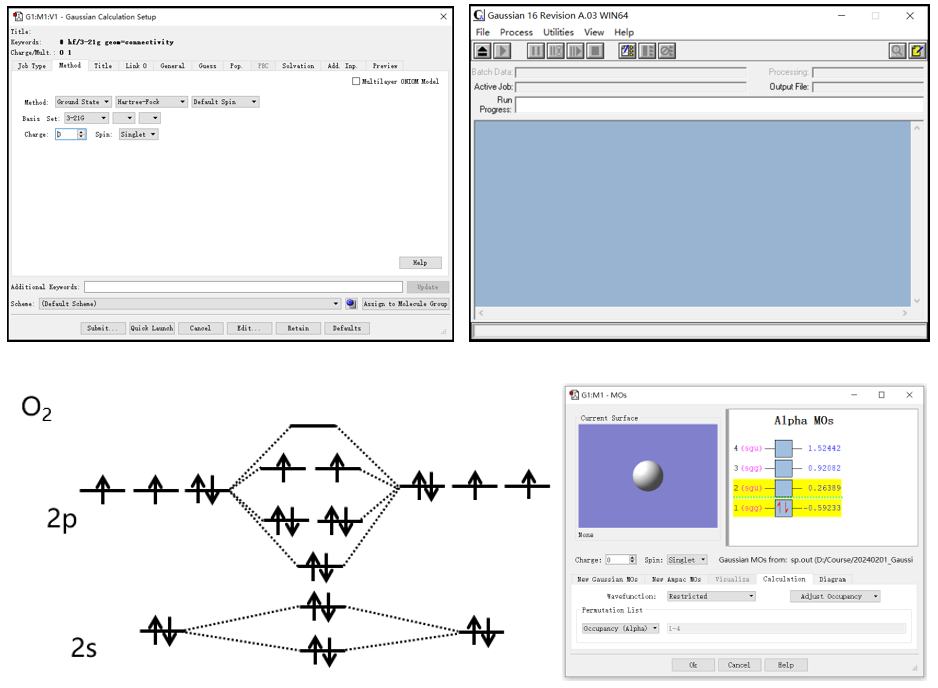
讲授Gaussian的基本计算,解读Gaussian的输入文件与输出文件,常用关键词,使用GaussView提交任务的方法。
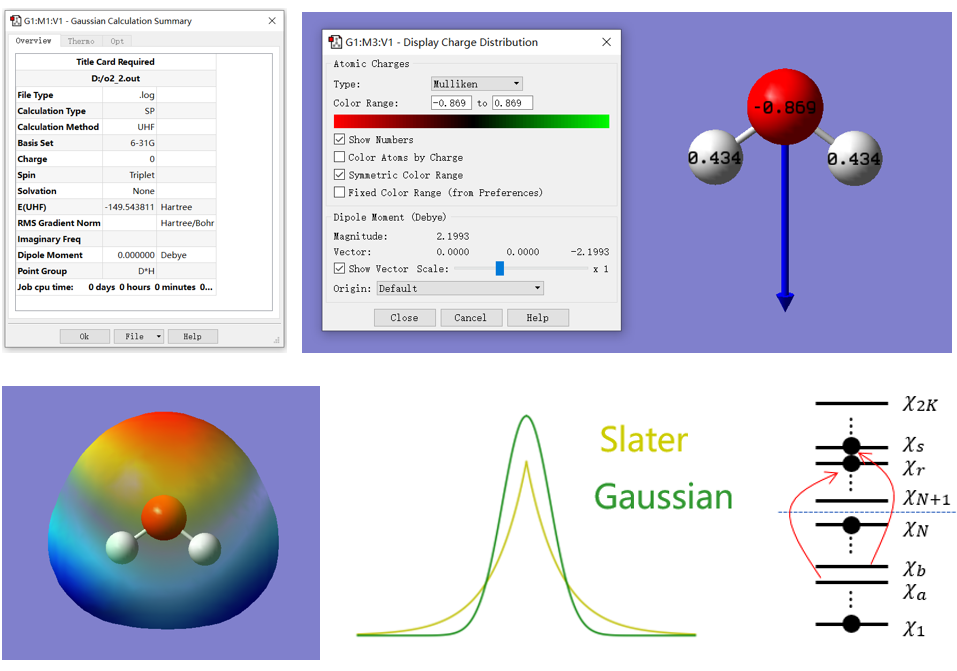
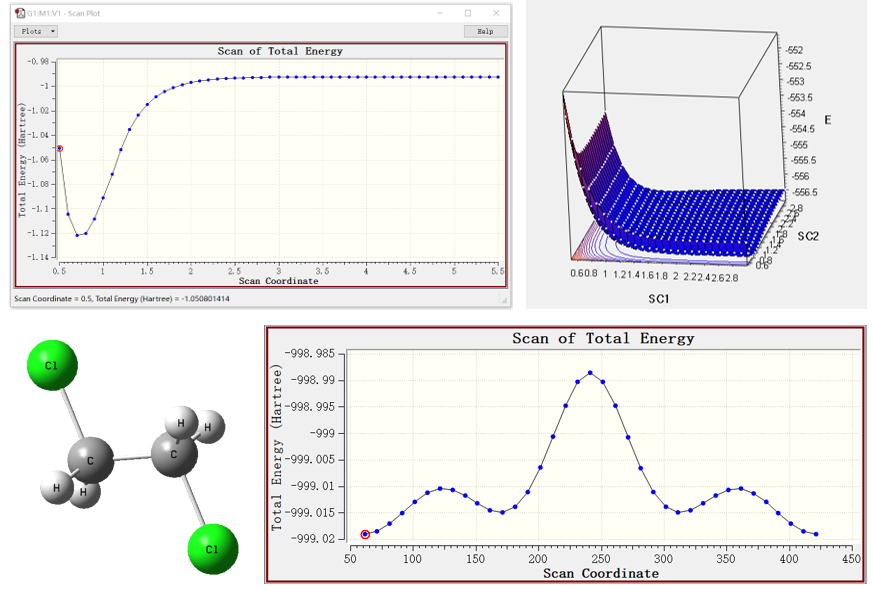
实操介绍单点能、几何优化、分子轨道、电荷布居、静电势、振动、零点能、热力学能、键解离能等常见计算。
学习Hatree-Fock极限与相关能概念,讲解后Hatree-Fock系列方法,包括组态相互作用方法、耦合簇方法、微扰方法等。
介绍size consistent与size extensive的概念。
讲解DFT常见泛函与选择,介绍组合方法,并对上述提到的方法进行实操演示。
学习Pople型基组、Dunning基组、赝势基组,并介绍基组的选择。
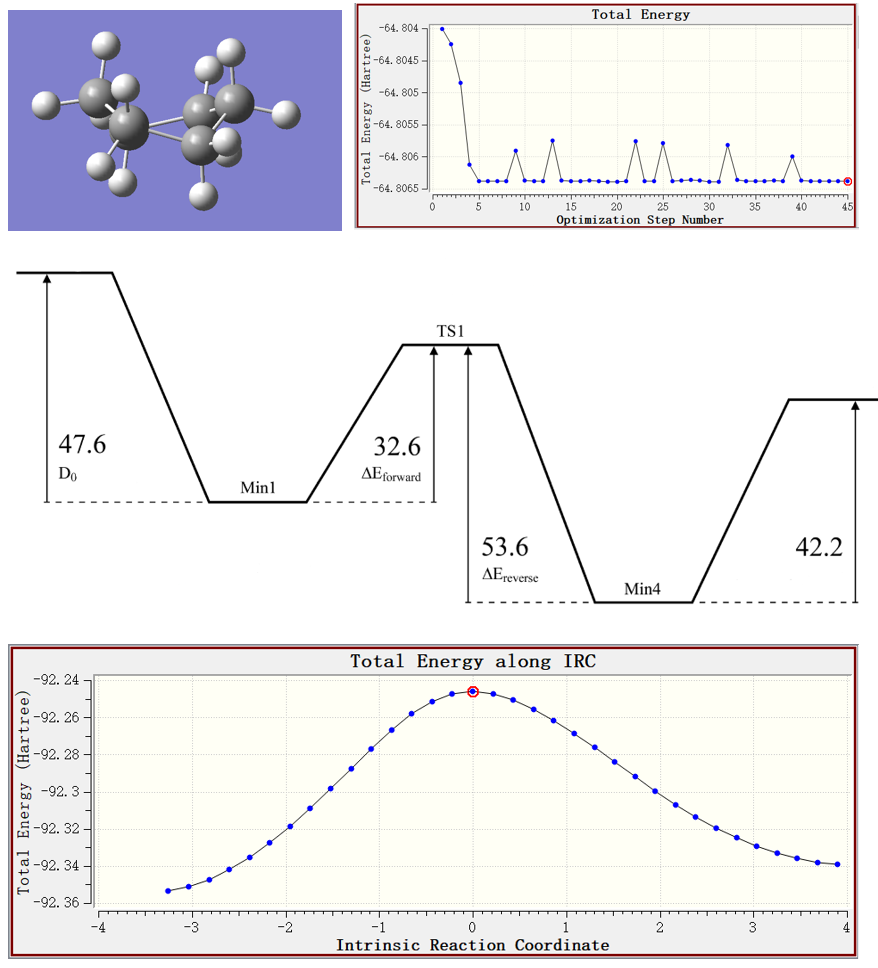
学习Gaussian计算过渡态的方法,实操TS方法、QST2方法的使用,并对两种方法进行对比。
学习IRC用途,并实操介绍在Gaussian中计算IRC的方法。
学习开壳层与闭壳层概念,对比RHF、UHF、ROHF方法。
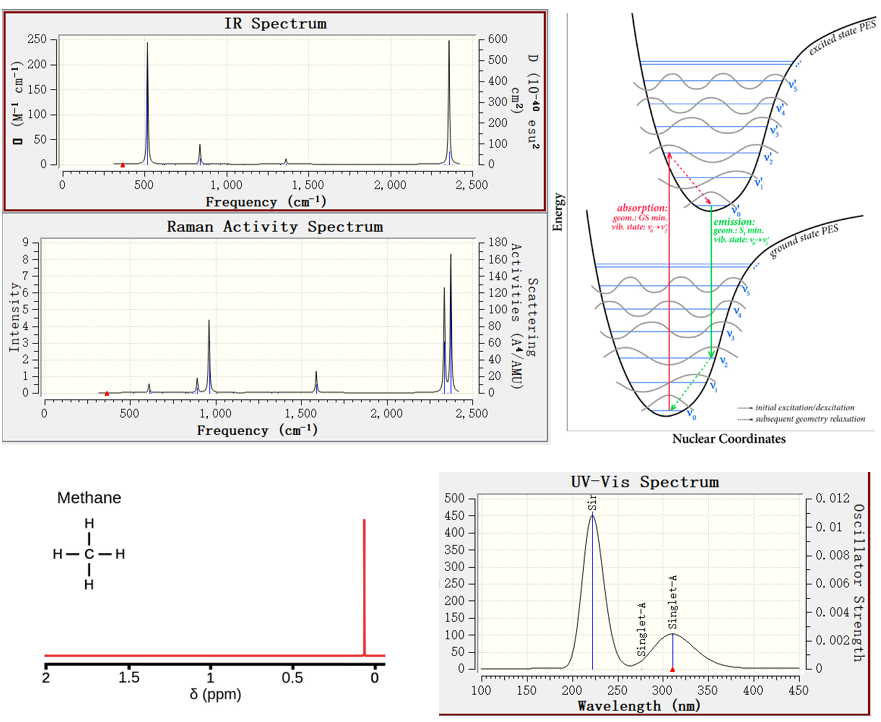
实操使用Gaussian计算红外光谱、拉曼光谱、紫外可见光谱、荧光光谱、磷光光谱、核磁共振谱的方法。
实操Gaussian同位素取代的计算,学习同位素取代对各计算的影响。
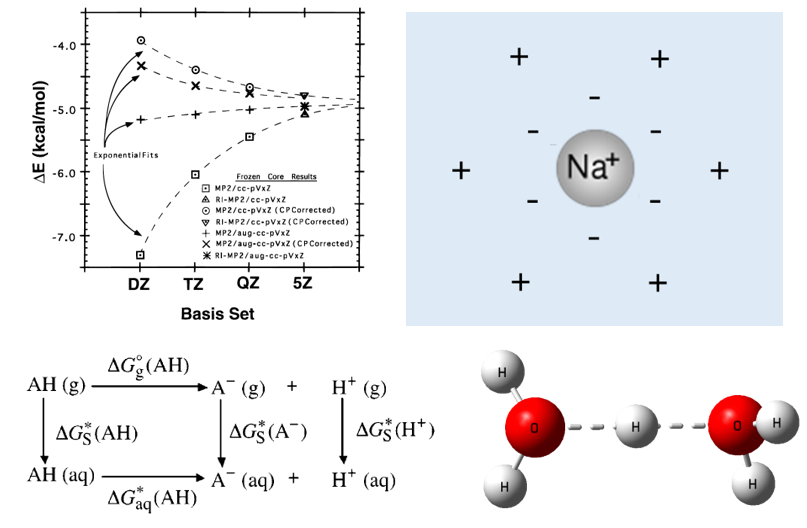
学习弱相互作用计算中的基组重叠误差错误,并结合实操学习解决办法。
学习使用PCM模型、SMD模型,实操学习溶解自由能的计算、pKa的计算。
主办单位:深圳华算科技有限公司(拥有VASP、Materials Studio、Gaussian、LAMMPS商业版权)
培训形式:线上课程,课程群永不解散,随时提问,及时解答。
课程费用:2980元,提供增值税普通发票及邀请函。老客户有优惠,请联系客服咨询。
报名方式:识别下方二维码报名,或者联系手机:133-1680-8231。
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!