

氢氧化反应(Hydrogen Oxidation Reaction, HOR)是氢燃料电池阳极的核心反应,其动力学缓慢性制约了低温燃料电池的效率。本文通过密度泛函理论(DFT)计算,系统解析HOR反应路径、活性描述符与催化剂设计策略,为突破催化瓶颈提供原子尺度解决方案!
一、HOR反应机理与关键步骤
HOR在酸性(PEMFC)和碱性(AEMFC)环境中的路径不同,以酸性条件为例:
1.H₂解离吸附:H₂(g) → 2H*(氢原子吸附,ΔG_H*为关键参数)
2.氢氧化步骤:H* → H⁺ + e⁻(Volmer步骤逆过程)
3.中间体耦合:H* + OH⁻→ H₂O(碱性条件涉及OH⁻参与)
决速步判定:
·酸性条件:H₂解离吸附(需低ΔG_H*)。
·碱性条件:OH⁻的吸附与反应(需优化OH*吸附能)。
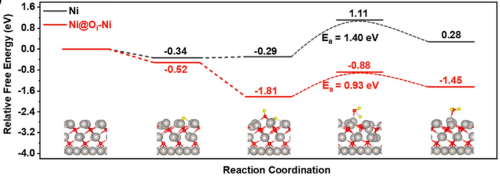

二、HOR与HER的对称性与差异
·热力学对称性:HOR与HER互为逆反应,理想催化剂需平衡双向活性。
·动力学差异:
oHOR在碱性条件下动力学更缓慢(需克服OH⁻+H+生成H2O能垒)。
oPt在酸性HOR中表现优异(ΔG_H* ≈ 0 eV),但在碱性中活性骤降。
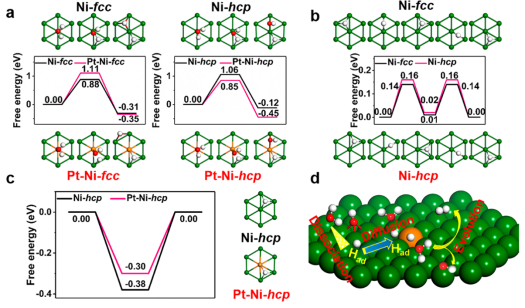
三、DFT计算HOR的核心参数
1. 氢吸附自由能(ΔG_H)*
·定义:H吸附态与气相H₂的自由能差,ΔG_H = ΔE_H* + ΔZPE – TΔS。
·理想值:ΔG_H* ≈ 0 eV(火山图顶点)。
2. 羟基吸附能(ΔG_OH)*
·碱性条件关键参数:OH*吸附过强阻碍反应位点,过弱则无法促进OH⁻耦合。
3. 活性火山图(Volcano Plot)
·横坐标:ΔG_H*或电子描述符(如d带中心ε_d)。
·纵坐标:交换电流密度(log(j₀))或活化能(E_a)。
4. 过电位(η)计算
·微观动力学模型:结合过渡态理论(TST)计算活化能垒。
·Butler-Volmer方程:关联过电位与电流密度。
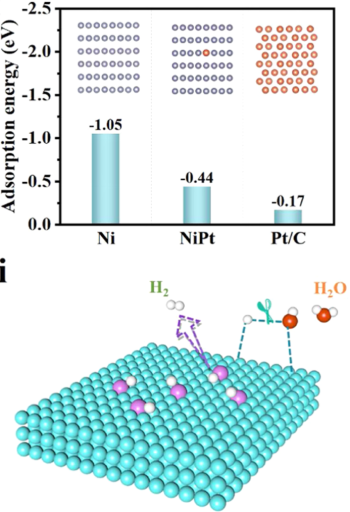
四、HOR催化剂DFT建模与计算流程
1. 模型构建
·表面模型:金属(Pt、Ni)或合金(PtRu)表面,优化晶胞参数。
·双位点设计:针对H与OH共吸附,构建双功能活性中心(如Pt-IrO₂界面)。
2. 计算参数设置
·泛函选择:
常规体系:GGA-PBE或RPBE(优化吸附能精度)。
含氧化物:PBE+U(U值参考文献,如IrO₂的U=3 eV)。
·溶剂化校正:
酸性条件:隐式溶剂模型(VASPsol)。
碱性条件:显式水层+OH⁻吸附(至少3层H₂O分子)。
3. 自由能校正
·振动频率计算:获取H与OH的零点能(ZPE)与熵变(TΔS)。
·电位与pH校正:
酸性:参考电极(RHE)电位U=0 V。
碱性:ΔG_OH需考虑pH依赖(ΔG_OH = ΔG_OH*° + k_BT ln(10) × pH)。
五、催化剂设计策略
1.双金属合金:
PtRu合金:Ru促进H₂解离,Pt优化H吸附(ΔG_H ≈ -0.05 eV)。
2.核壳结构:
Pd@Pt核壳:表面应变调控d带中心,平衡HOR/HER活性。
3.单原子催化剂(SACs):
Pt₁/CeO₂:载体氧空位促进OH⁻吸附,提升碱性HOR活性。
4.氧化物-金属界面:
Pt/Ni(OH)₂:Ni(OH)₂提供OH⁻吸附位点,Pt促进H₂解离。
益于理论计算化学的快速发展,计算模拟在纳米材料研究中的运用日益广泛而深入。科研领域已经逐步形成了“精准制备-理论模拟-先进表征”的研究模式,而正是这种实验和计算模拟的联合佐证,更加增添了论文的可靠性和严谨性,往往能够得到更广泛的认可。
华算科技已向国内外1000多家高校/科研单位提供了超过50000项理论计算和测试表征服务,部分计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。
