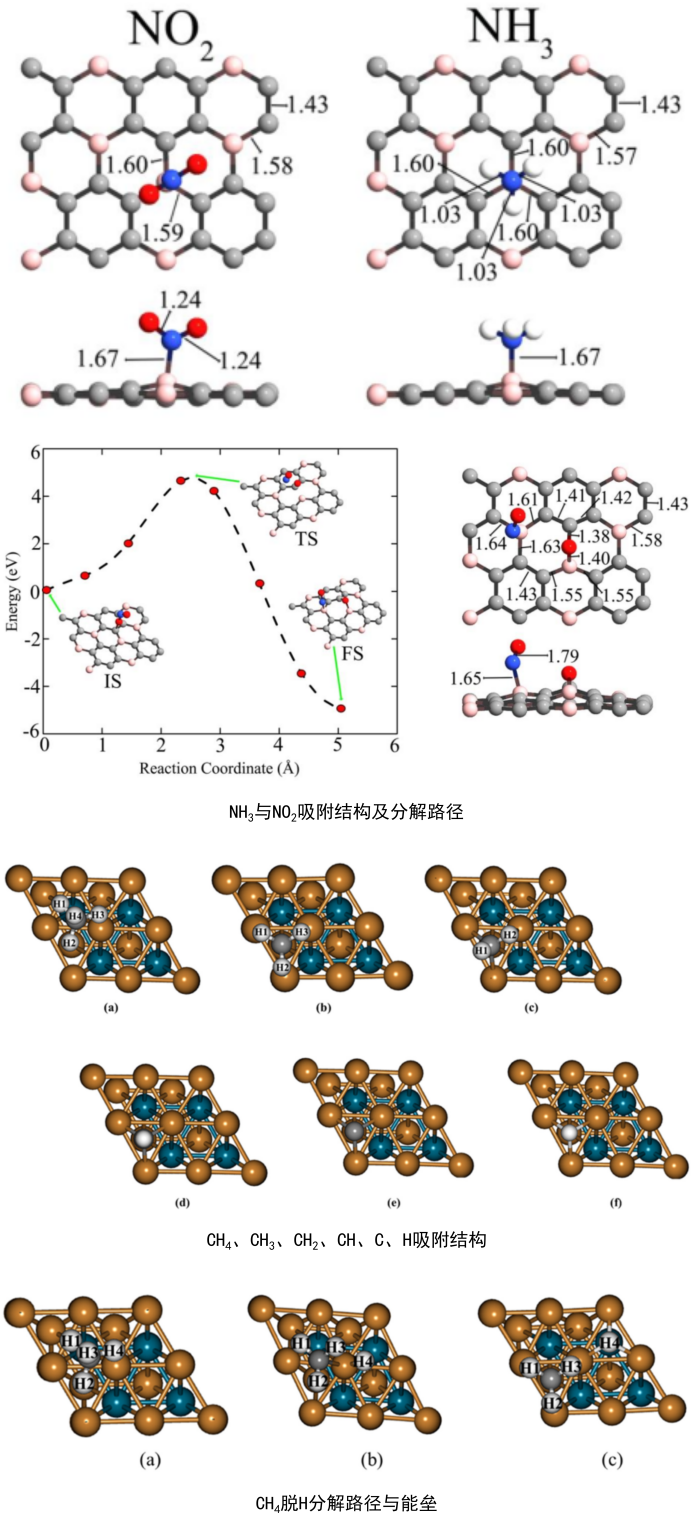
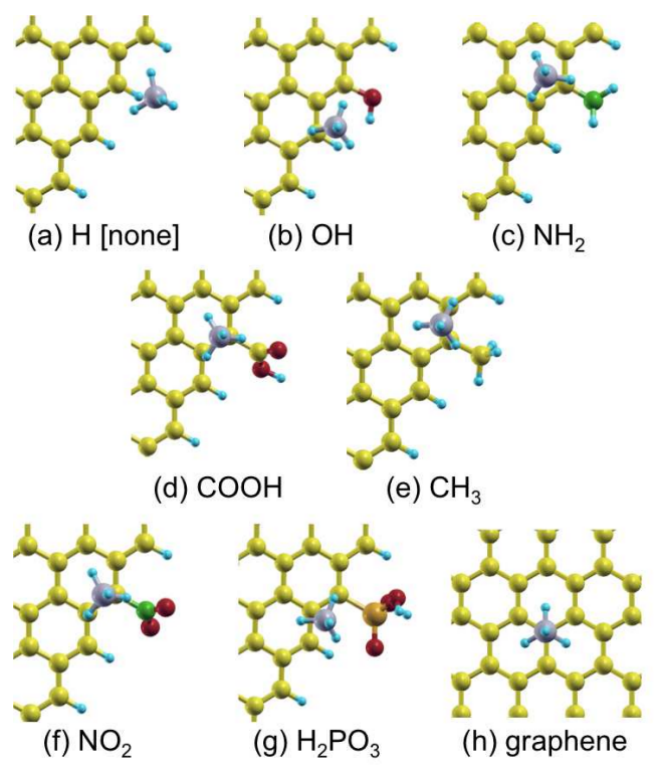
想要获得各种分子在晶体表面、二维材料、一维材料表面活性位点的吸附构型与吸附能吗?
想要将常规的三维催化剂结构转变为可用于计算的表面与二维结构吗?
本次课程由华算科技技术总监朱老师主讲材料表面吸附和催化性质,DFT计算软件选择专业技术人员一致推崇的VASP。实操计算边学边练,尽早学习更早受益!
具体涉及:晶体表面/二维结构/一维结构的结构性质,功函数、d带中心、吸附能、差分电荷密度等电子性质,HER、OER/ORR、NRR、CO2RR等催化反应的自由能与过渡态计算,CO、CO2、CH4、C6H6等分子的吸附与分解过程(课表详见后文)。
目前已上架小鹅通,并建立专属课程群,实时答疑。随时随地永久学,边学边练搞定吸附与催化计算!我们会根据学员反馈,不断精选和更新课程内容,一次购买,持续增值!
报名成功会同时赠送价值2400元的三套量子化学基础、二维材料计算、铁电计算课程。
报名方式:识别下方二维码报名,或者联系手机133-1680-8231。
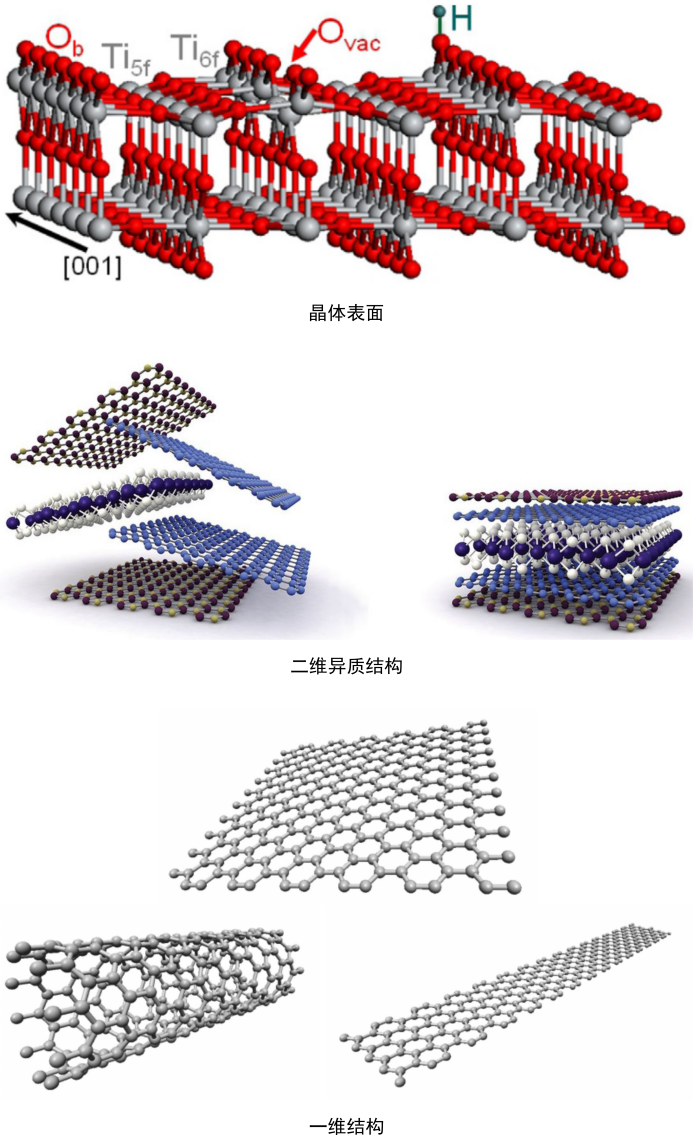
结构性质计算:晶体结构,晶体表面结构、二维结构、掺杂二维结构、一维结构、二维异质结构、无磁、铁磁、反铁磁、亚铁磁、结构优化、层间距、原子磁矩;
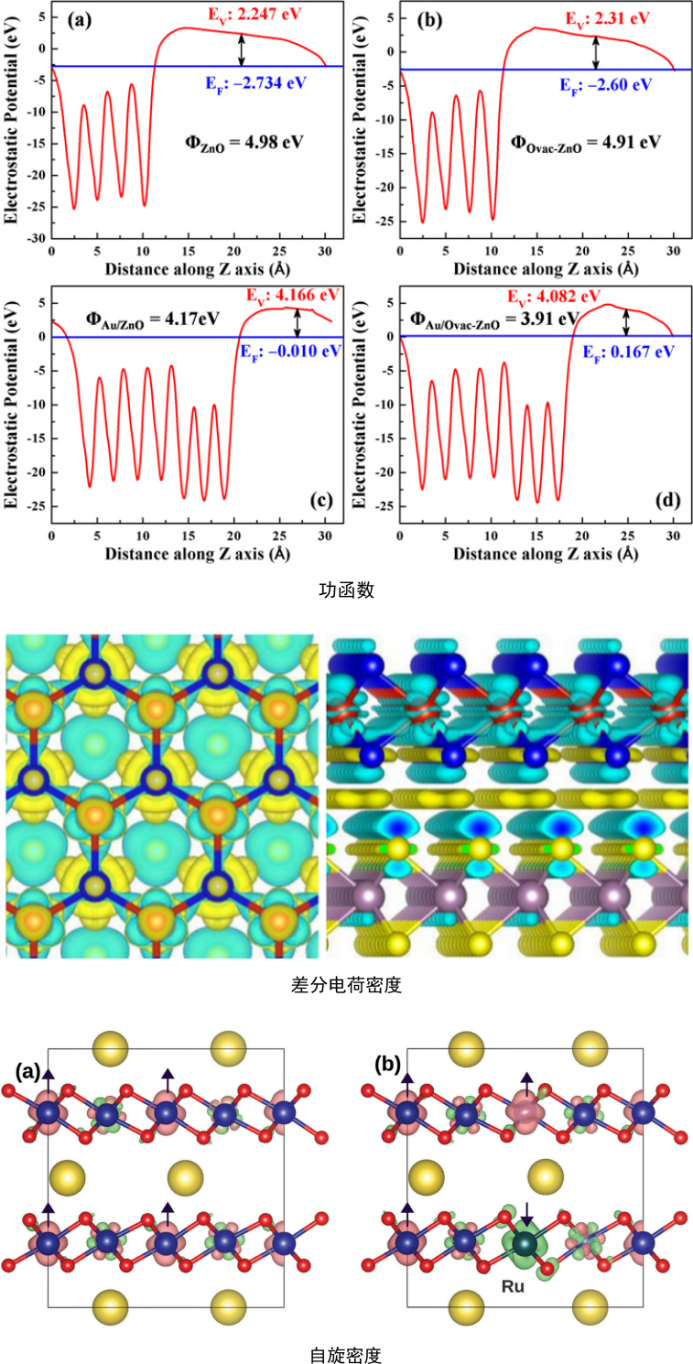
电子性质计算:电荷密度、自旋密度、差分电荷密度、结合能、静电势、功函数、态密度、d带中心;
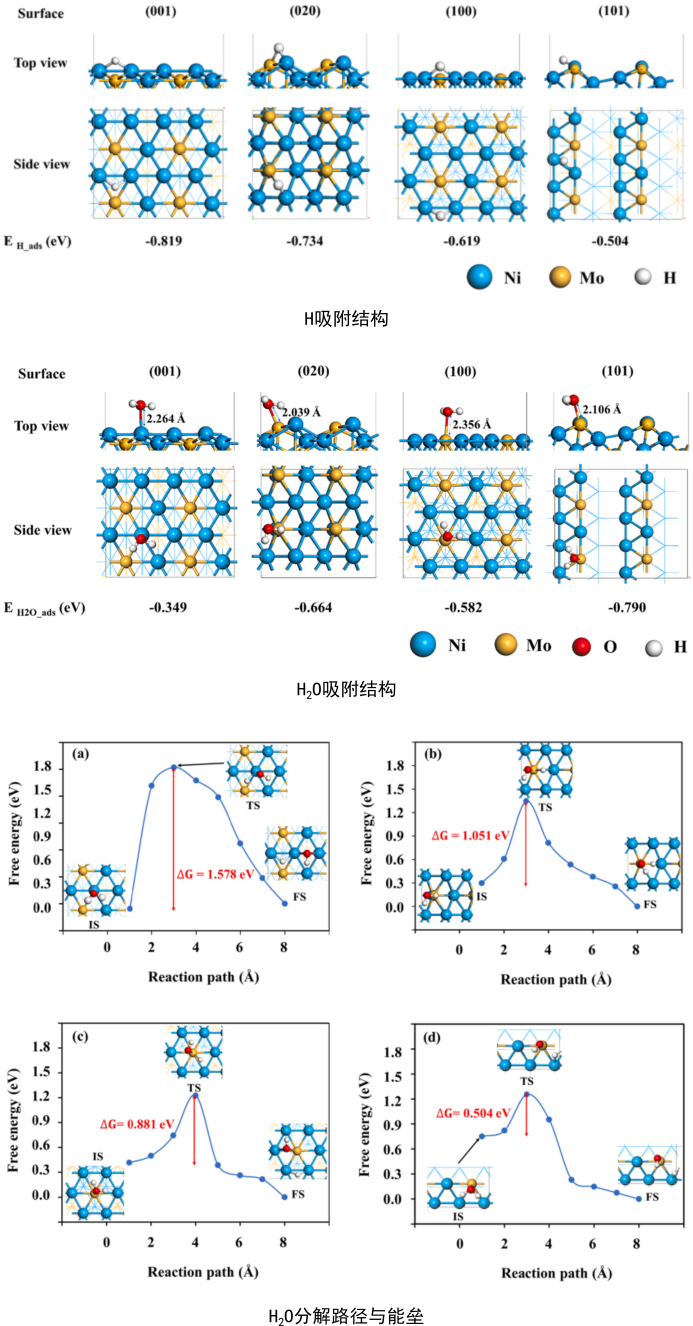
HER:反应路径、反应位点、H吸附、H-OH吸附、H2O吸附、零点振动能、熵、自由能、HER台阶图、H2O分解反应能垒与路径、虚频、虚频消除;
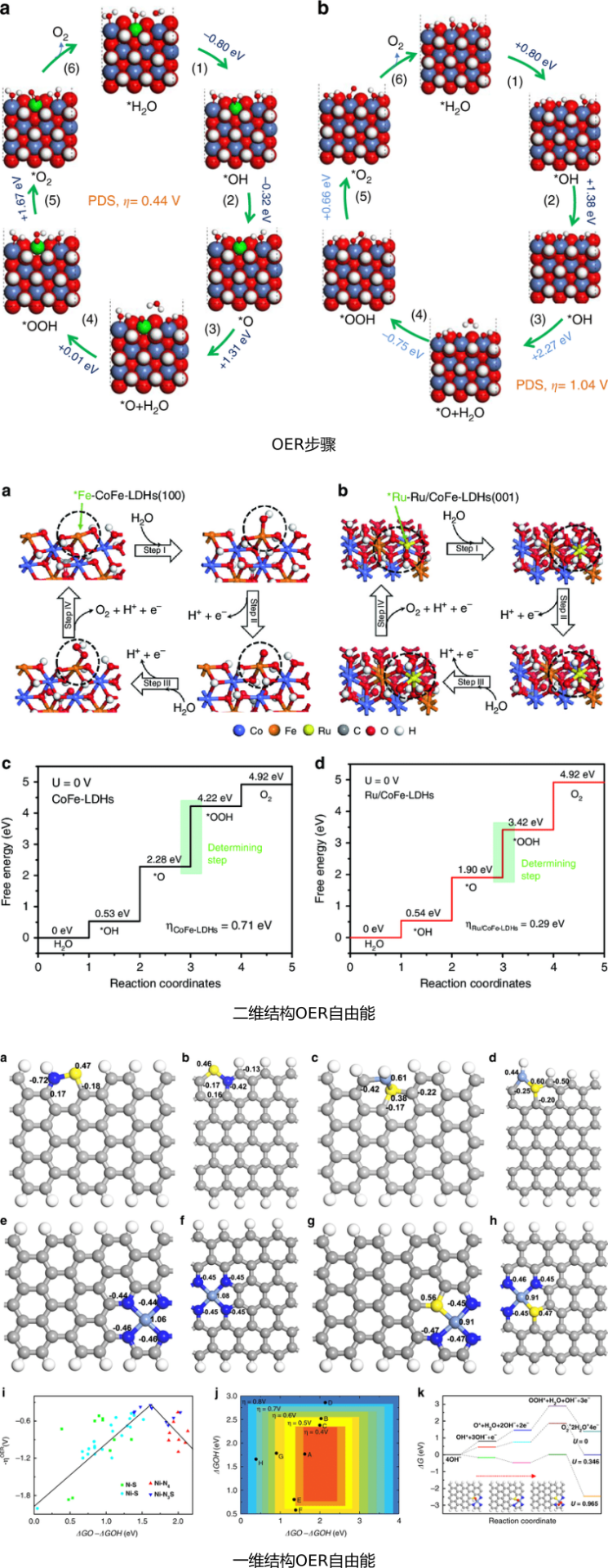
OER:反应路径、反应位点、H吸附、OH吸附、OOH吸附、零点振动能、熵、自由能、OER/ORR台阶图、OOH脱H反应能垒与路径;
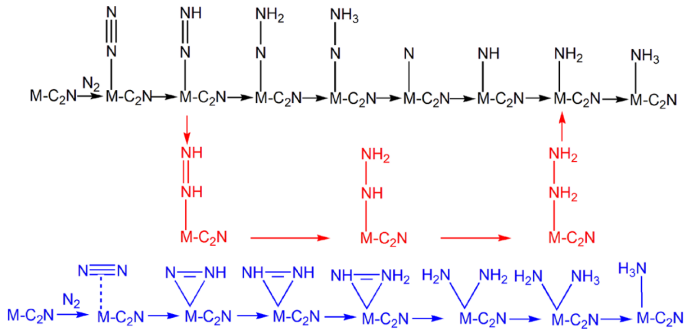
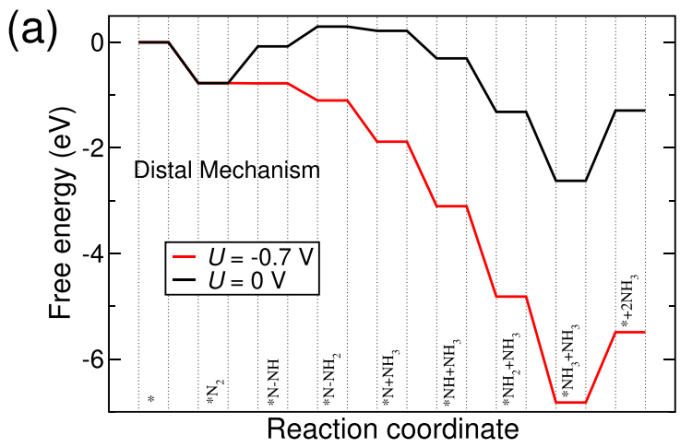
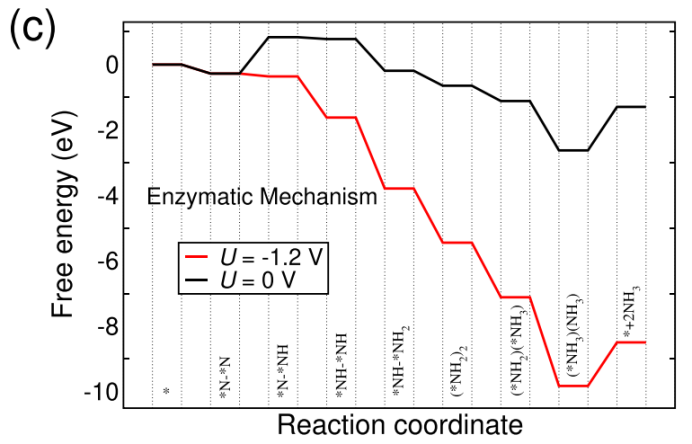
NRR:反应路径,Distal路径、Consecutive路径、Alternating路径、Enzymatic路径,中间体结构优化、零点振动能、熵、自由能、NRR台阶图;
CO2RR:反应路径、反应位点、COOH吸附、CO吸附、零点振动能、熵、自由能、CO2RR自由能计算、电压对自由能影响、催化剂线性约束关系;
吸附性质计算:NH3、CH4、CH3F、CH2F2、CHF3、CF4、C6H6吸附,C吸附、O吸附、CO吸附、CO2吸附,CH、CH2、CH3、CH4脱H反应能垒与路径,CO分解为C和O反应能垒与路径、CO2分解为CO和O反应能垒与路径。
注意:本次课程为VASP催化专题课程,需要有一定的VASP计算基础。零基础学员建议同时报名VASP计算零基础入门培训:晶体结构、电子、弹性、光学、磁性、电池、催化性质计算(点击文字可查看课程详情)。
朱老师,同济大学本科直接攻读博士学位(4年),海外3年以上博后经历,发表高质量独立一作SCI论文30篇,回国后被授予深圳市海外高层次人才,拥有14年VASP重度使用经验,成功讲授100+场VASP计算培训和超过10W人的学习理论计算公开课。
课程内容包括晶体表面/二维结构/一维结构的结构性质,功函数、d带中心、吸附能、差分电荷密度等电子性质,HER、OER/ORR、NRR、CO2RR等催化反应的自由能与过渡态计算,CO、CO2、CH4、C6H6等分子的吸附与分解过程
使用开源的VESTA与ASE软件构建晶体表面(slab)模型、掺杂与未掺杂二维结构、二维异质结构、一维结构模型。计算结构性质,包括面间距与原子磁矩。这能够让大家熟练掌握模型构建方法与软件操作技巧,为后续计算提供结构模型。
晶体表面静电势、功函数、d带中心计算,掺杂对二维结构功函数影响计算,二维异质结构差分电荷密度、结合能计算,一维结构电荷密度、自旋密度计算。这能够让大家掌握吸附剂与催化剂材料的基本电子性质,为后续反应中吸附位点判断提供参考依据。
金属是一类常见的催化剂材料,催化反应发生在表面的某些活性位点上。HER涉及到催化剂对H2O、H、H-OH中间体的吸附以及H2O的分解过程。计算HER中间体的自由能(包括总能、零点振动能、熵)、H2O分解的路径与能垒(包括过渡态与虚频),并绘制HER的台阶图。
金属氧化物是一类理想的催化剂,OER/ORR涉及中间体OH、O、OOH的吸附。计算中间体的自由能(包括总能、零点振动能、熵)、OOH脱H反应动力学过程,并绘制OER/ORR台阶图。
过渡金属原子掺杂是调控催化剂活性的一种手段,NRR主要涉及4条反应路径(Distal、Alternating、Enzymatic、Consecutive)。计算不同反应路线中间体的自由能(包括总能、零点振动能、熵),绘制NRR台阶图,并判断反应路线发生的可能性。
NRR路径与中间体结构
NRR路径与自由能
电压是调控电化学反应势垒的常见手段。CO2RR涉及中间体COOH、CO等中间体的吸附。计算中间体的自由能(包括总能、零点振动能、熵)以及电压对自由能的影响,并绘制CO2RR台阶图。
NH3、CH4、CH3、CH2、CH、C6H6吸附位点与分子空间构型的筛选与计算,对比掺杂与未掺杂位点的吸附效果,CH4分解为CH3和H的迁移路径与能垒计算,CH3分解为CH2和H的迁移路径与能垒计算,CH2分解为CH和H的迁移路径与能垒计算,CH分解为C和H的迁移路径与能垒计算。这能够让大家理解掺杂与未掺杂位点的吸附效果差别,掌握基于C和H的基团与分子吸附、分解、迁移的计算技巧。
C、O、CO、CO2吸附位点与分子空间构型的筛选与计算,对比边缘与体相位点的吸附效果,CO分解为O和C的迁移路径与能垒计算,CO2分解为CO和O的迁移路径与能垒计算。这能够让大家理解体相与边缘位点的吸附效果差别,掌握基于C和O的基团与分子吸附、分解、迁移的计算技巧。
CO2分解反应路径
主办单位:深圳华算科技有限公司(拥有VASP、Materials Studio、Gaussian商业版权)
培训形式:录播课程,提供课件、超算练习账号,课程群永不解散,在线答疑。
课程费用:2980元,限时赠送价值2400元理论计算课程(包含价值699元量子化学理论入门宝典《计算量子化学入门必备线上课程》、价值799元《二维材料铁电性计算入门课程》、价值899元《经典二维材料理论计算课程》。
报名方式:识别下方二维码报名,或者联系手机133-1680-8231。
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!