



与传统的热催化方法相比,直接电化学乙烯转化为乙二醇(C2H4-to-EG)可以潜在地减少化石燃料的消耗和二氧化碳(CO2)的排放。电沉积法制备钯(Pd)是一种很有前途的电催化剂;然而,该工艺存在着乙二醇电流密度(EG)较低(-2),法拉第效率(-1),阻碍了实际应用。
清华大学段昊泓、李必杰等人报道了一种负载在大面积气体扩散电极上的纳米枝晶钯催化剂。该催化剂具有较高的EG电流密度(12 mA cm-2)和产率(227 μmol h-1),但法拉第效率较低(65%)。随着Cl–离子的进一步修饰,法拉第效率提高到92%的历史最高值,EG电流密度(18 mA cm-2)和生产率(~340 μmol h-1)也得到了提高。
实验数据表明,Cl–的强吸电子特性降低了原位生成的Pd-OH的氧化能力,抑制了EG对乙二醇醛的过度氧化。同时,Cl–改变了EG在Pd表面的吸附构型,从平行和双位配位变为垂直和单位配位,从而阻止了EG对CO2的C-C键裂解。此外,Cl–吸附有利于Pd-OH活性物质的生成,提高了催化活性。这项工作证明了表面离子修饰在提高C2H4-to-EG直接电化学转化的活性和选择性方面的巨大潜力,这可能对多种增值化学品的电合成具有重要意义。
相关工作以《Direct Electrooxidation of Ethylene to Ethylene Glycol over 90% Faradaic Efficiency Enabled by Cl– Modification of the Pd Surface》为题在《Journal of the American Chemical Society》上发表论文。这也是段昊泓副教授在《Journal of the American Chemical Society》上发表的第9篇论文。

段昊泓博士,国家杰出青年科学基金获得者,国家重点研发计划首席科学家。研究兴趣包括纳米催化和电催化。近几年,致力于解决塑料、生物质等碳资源的化学循环和高值化利用问题,发展电/光驱动的绿色催化方法,基于对反应机理的基础研究发展新型电催化剂和高效反应装置,在温和条件下将废弃塑料和生物质等碳资源转化为高附加值化学品,并将上述反应与电解水制氢、二氧化碳还原过程耦合,对推动化学品绿色合成和碳中和具有重要意义。研究成果以第一作者或通讯作者发表在 Nat. Catal.、Nat. Commun.、J. Am. Chem. Soc.、Angew. Chem. Int. Ed.、Chem. Soc. Rev. 等国际学术期刊。
李必杰,2003年进入北京大学化学学院学习,2007年毕业获得学士学位。同年进入北京大学化学学院施章杰教授课题组攻读博士学位,进行金属有机化学方向的研究工作,并于2012年获得博士学位。2012年8月加入美国加州大学伯克利分校Hartwig实验室开展博士后研究,博士后期间的工作集中在过渡金属催化反应的机理研究。2015年8月进入清华大学基础分子科学中心开展独立研究工作。2020年获基金委优秀青年科学基金资助。研究方向是金属有机化学、烯烃不对称催化、资源有机电化学。
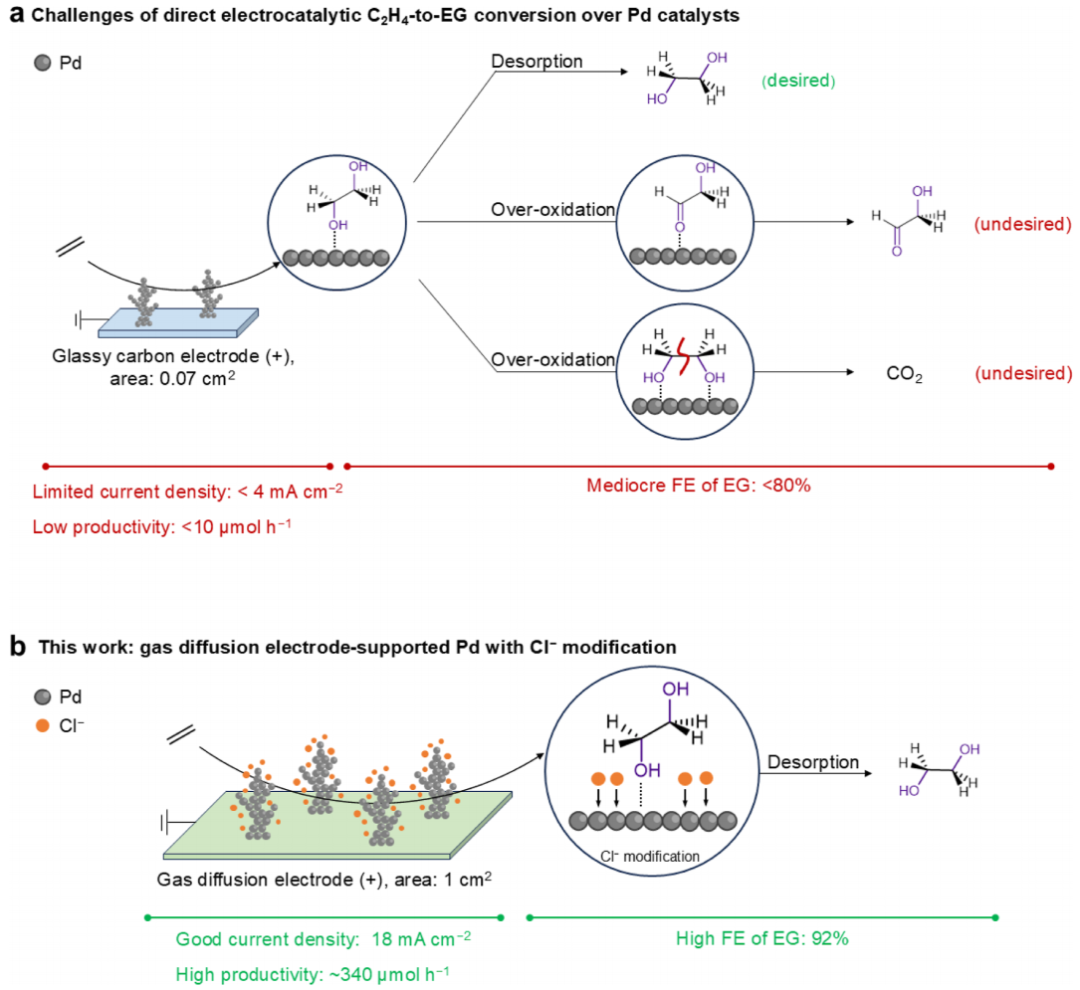
图1 C2H4氧化的反应途径
对于Pd基催化剂,产生活性羟基(OH*)的水活化能力对jEG的影响很大。通过在Pd中掺杂Au,可以精细调节OH*的结合能,降低OH加成C2H4的活化势垒。然而,jEG仅有一定程度的增加(从~ 3到~ 4 mA cm-2;图1a左)。为了增加FEEG(图1a右),关键是增强所需的C2H4形成(上途径),同时抑制法拉第副反应,包括EG过度氧化为乙醇醛(GA;中间途径)和二氧化碳(底部途径),以及C2H4水化,然后氧化为乙醛(AA)和乙酸(AcOH)。特别是,在Pd上,EG产生的CO2非常严重,因为EG倾向于在Pd表面平行吸收,从而导致C-C键裂解形成CO2。因此,很少观察到FEEG>80%的钯催化剂。此外,由于使用工作面积较小(0.07 cm2)的玻碳电极来负载Pd,因此整体EG生产率(-1)远不能实现。总的来说,开发具有高电流密度、高FE和生产率的高效Pd催化剂,以实现C2H4到EG的直接电化学转化,仍然是一项艰巨的挑战。
本文报道了一种经Cl–吸附修饰的Pd催化剂,在C2H4的直接电化学氧化中表现出很高的催化性能(图1b)。具体而言,以气体扩散电极(GDE)(工作面积为1 cm2)为载体,采用电沉积法合成了Pd纳米枝晶催化剂。电解质中Cl–对提高催化性能相当重要。经过Cl-吸附后的Pd,观察到jEG(10~12 mA cm-2)、FEEG(56%~65%)和EG产率(175~227 μmol h-1)的变化。
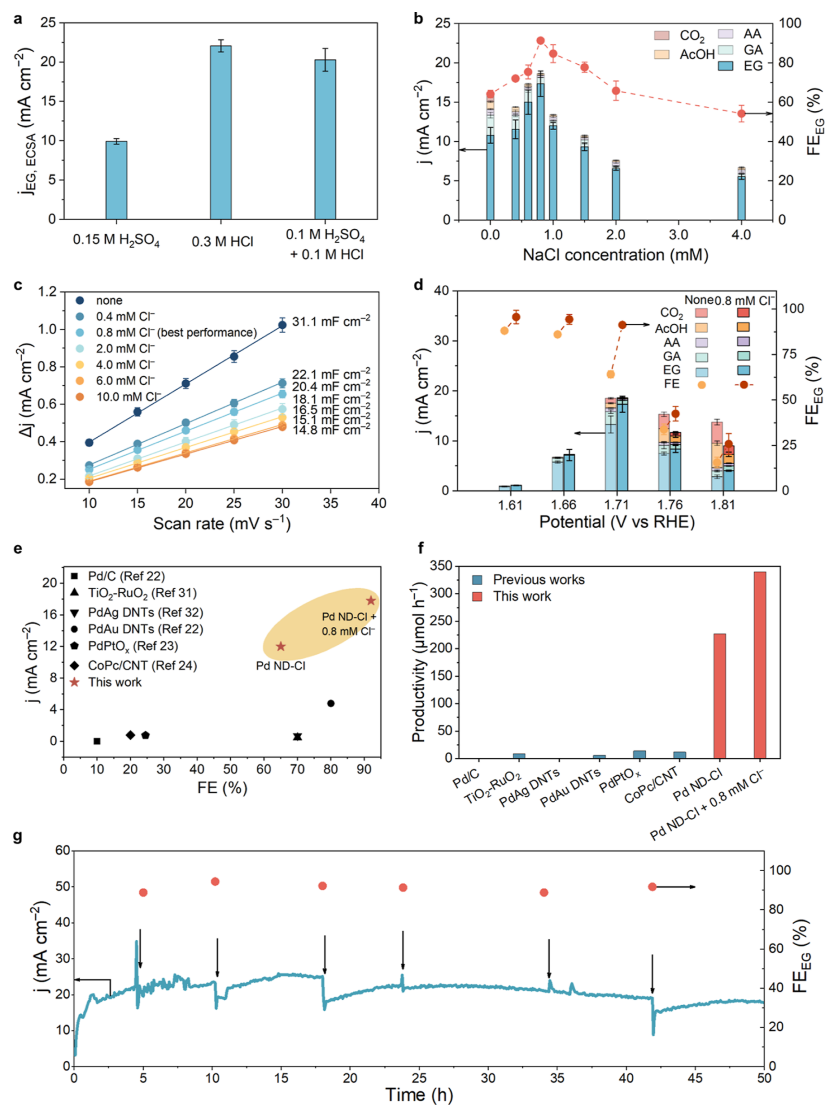
图2 C2H4氧化的电化学性能
ECSA归一化jEG(jEG,ECSA)值表明电沉积在HCl中的Pd具有较高的电流密度(20 mA cm-2),电沉积电解质为HCl>H2SO4和HCl混合物>H2SO4(图2a)。因此,推测Cl–的存在可能有助于促进jEG,ECSA和FEEG。考虑到在钯催化剂合成过程中引入Cl–的积极作用,预计在电解液中加入Cl–进行电化学C2H4氧化也可能产生积极的催化效果。为了证明这种可能性,使用Pd ND-Cl作为催化剂,在含有不同NaCl浓度(0 ~ 4.0 mM)的0.1 M NaClO4中进行了电化学氧化C2H4。如图2b所示,当添加0.8 mM NaCl时,与不添加NaCl或添加其他NaCl浓度的Pd相比,Pd的催化性能显著提高(jEG: 17.4 mA cm-2,FEEG:92%,EG产率:340 μmol h-1)。
NaCl浓度的进一步增加导致jEG和FEEG的降低,这可能是由于过量的Cl–阻断了催化剂上的活性位点。当引入超高浓度的NaCl(100 mM)时,几乎没有观察到电流密度。通过测量不同NaCl浓度下Pd ND-Cl样品的ECSA(图2c),观察到ECSA持续下降,验证了Cl–吸附可能阻断Pd表面。此外,当采用较大的NaCl浓度时,Pd上的Cl–覆盖率更高。结果,当存在0.8 mM NaCl时,估计Cl–覆盖率为~ 59%。当NaCl浓度低于0.8 mM时,Pd ND-Cl的jEG、ECSA得到改善;结果表明,吸附Cl–促进了Pd ND的固有活性。在1.6~1.8 V范围内,Cl–对jEG和FEEG的促进作用是有效的(图2d)。值得注意的是,尽管jEG较低,但在相对较低的电位(1.61和1.66 V)下,FEEG可以达到约95%。在较高电位(1.76和1.81 V)下,过氧化产物的FEs也受到Cl–存在的抑制。但由于Pd钝化作用,jEG较1.71 V时有所下降。
总的来说,在最佳条件下(电解液:0.1 M NaClO4+0.8 mM NaCl;应用电位:1.7 V),良好的催化性能(jEG:17.4 mA cm-2,FEEG:92%)显示在Cl–修饰的Pd催化剂上,比以前的催化剂更有利(图2e),包括TiO2-RuO2催化剂,Pd基催化剂和CoPc/CNT催化剂。此外,由于使用GDE作为Pd的载体,具有较大的面积和Cl–修饰Pd的固有高活性,与先前的结果相比,达到了30倍的EG生产率(340 μmol h-1)(图2f),表明其潜在的应用前景。通过电解50 h评估了Cl–对提高催化性能的稳定性(图2g)。在长期电解过程中,平均jEG为18.0 mA cm-2,FEEG为91.8%,没有明显的催化性能损失。
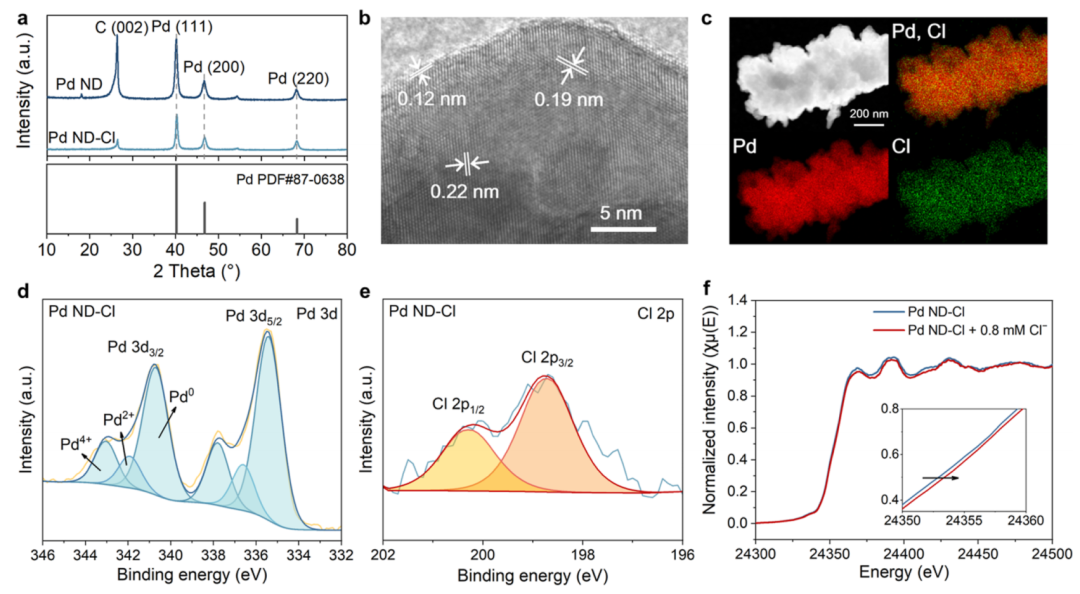
图3 Pd ND-Cl催化剂的表征
XRD谱表明,Pd ND和Pd ND-Cl均呈现面心立方晶体结构,暴露于(111)、(200)和(220)晶面(图3a)。HRTEM图像(图3b)显示,Pd ND-Cl主要暴露(111)、(200)和(220)面,对应不同晶格间距分别为0.22、0.19和0.12 nm,表明Cl–可能没有结合到Pd的主体中。EDS结果显示,Cl物质均匀分布在Pd表面(图3c)。
为了研究Cl–对Pd电子结构的影响,采用了XPS。如图3d所示,Pd ND(没有Cl–吸附)的Pd 3d5/2信号可以拟合成三个峰,分别位于335.1 eV(Pd0)、336.2 eV(Pd2+)和337.8 eV(Pd4+)。相比之下,Pd ND-Cl的这些峰呈现出正位移,这意味着由于Pd具有很强的吸电子能力,在Cl–存在的情况下,Pd容易失去电子。此外,Pd ND-Cl的Cl 2p的XPS谱中还可以观察到Cl 2p3/2峰(198.5 eV)和2p1/2峰(200.5 eV),表明氯离子以Cl–的形式存在于Pd表面(图3e)。
在开路电位(OCP)条件下,对0.1 M NaClO4进行了原位XAS测量。Pd ND-Cl的归一化Pd的L3边缘XANES谱图(图3f)显示,在电解质中加入0.8 mM NaCl后,吸附边向高能位置移动,说明发生了Pd向Cl–的电子转移。这些结果与XPS结果一起表明,Pd表面的Cl–吸附调节了Pd的电子结构,这可能会改变Pd在C2H4氧化中的电化学氧化反应性。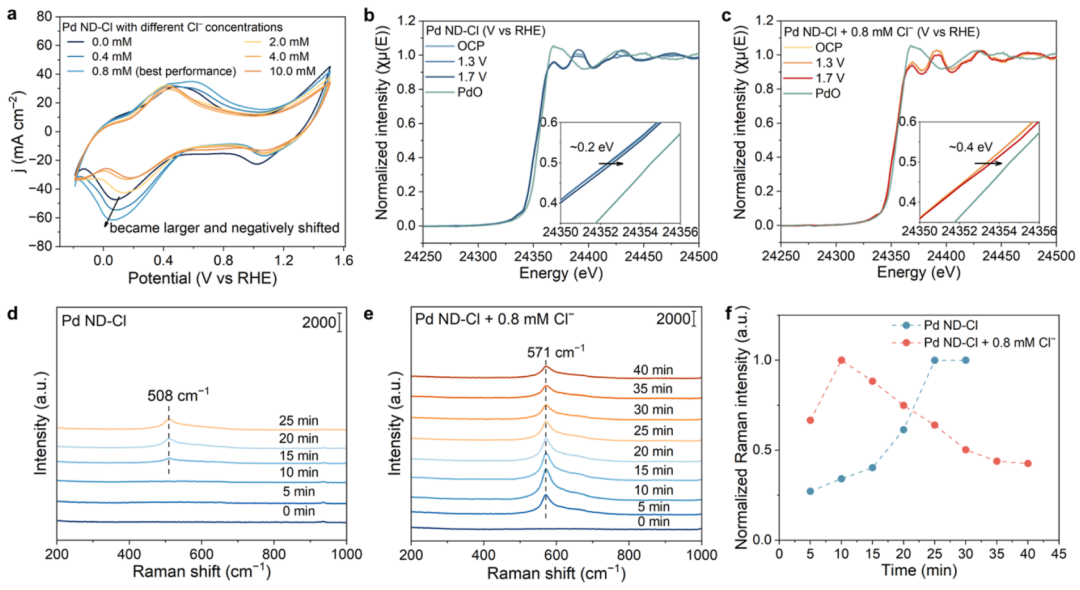
图4 水活化过程的机理研究
根据之前的报道,C2H4-to-EG在Pd催化剂上的转化可能是通过Pd上的水活化生成Pd-OH,随后连续添加C2H4。因此,研究了Cl–吸附对Pd-OH生成的可能影响。通过CV测量来探测有或没有NaCl的Pd-OH物质的生成(图4a)。在Pd ND上,0.08 V的还原峰可以分配给PdO和表面吸收OH(OH*)的还原;即Pd-OH物种)。随着NaCl浓度的增加,还原峰强度增加并发生了阴极位移,这反映了Cl–的存在促进了Pd-OH的生成,从而促进了水活化过程,这与Pd ND-Cl增加的jEG一致(图3b)。然而,当NaCl浓度超过0.8 mM时,观察到还原峰的反向变化(强度下降,阳极偏移),表明Pd-OH的生成受到抑制,可能是由于Pd表面被Cl–阻断,这与大量NaCl加入时jEG的减少一致(图3b)。
上述XPS(图3d、e)和XANES(图3f)结果表明,Pd发生了电子向Cl–的转移;因此,有理由认为Pd更容易被水活化氧化,形成Pd-OH。为了深入了解电化学条件下Cl–加入后催化剂的电子结构变化,进行了原位XAS测量。从归一化Pd的L3边缘XANES光谱(图4b)中可以看出,在没有Cl–的情况下,在1.3 V的施加电位下,吸收边缘保持不变,这与该低电位下不发生反应的电化学性能是一致的。当外加电位为1.7 V时,吸收边向能量更高的位置移动,表明Pd的价态增加。在Cl–存在下(加入0.8 mM NaCl;图4c),吸收边更大程度地向高能位置移动。具体来说,在不添加Cl–的情况下,在1.7 V条件下,归一化强度为1.1时,吸收边只偏移了~ 0.2 eV,而在相同条件下,当Cl–存在时,吸收边偏移了~ 0.4 eV。
对GA的抑制作用(FE为6%~2%)优于氧化能力较弱的催化剂。为了评估Pd ND和Pd ND- Cl产生的Pd-OH物种的差异,进行了原位拉曼测量(图4d、e)。在施加1.7 V的电位时,出现了以508 cm-1为中心的特征性宽峰,这可以分配给Pd-OH的信号,与先前的报道一致当加入0.8 mM NaCl时,特征峰蓝移至571 cm-1,强度变强(图4e),表明Pd-OH结合更强,从而降低OH*的氧化能力,抑制过氧化。此外,Pd ND-Cl的峰值强度增加更快,这意味着水活化更容易,这与前面的CV结果一致(图4a)。10 min后峰值强度下降,达到稳定状态(图4f),初步认为这是由于Cl–的竞争性吸附,释放了表面吸附的OH。
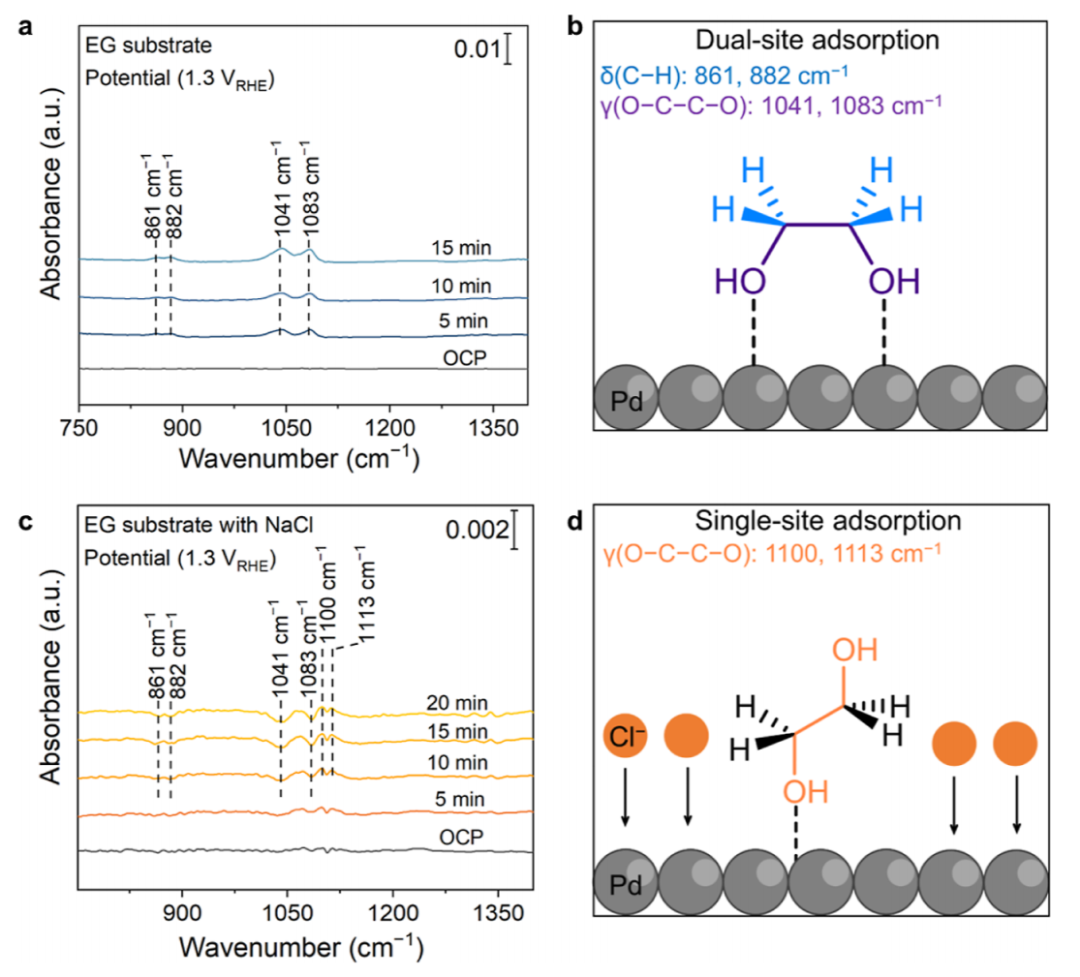
图5 机理研究
为了研究EG在Pd表面的吸附构型以及Cl–的相关影响,采用了原位衰减全折射-傅里叶变换红外光谱法。在测量过程中,以EG作为基底,施加1.3 V的阳极电位来加强EG在Pd表面的吸附。在没有NaCl的情况下(图5a),在861、882、1041和1083 cm-1处观察到几个特征峰。前两个峰属于CH2弯曲振动,后两个峰属于O-C-C-O的拉伸振动模式(图5b),共同决定了EG在Pd ND上的双位点(两个OH)配位平行吸附。相比之下,在NaCl的作用下(图5c),四个特征峰的强度都有所下降,而在1100和1113 cm-1处出现了另外两个峰。这两个峰被分配到O-C-C-O的不同拉伸振动模式,表明EG的结构改变为与其单一OH基团的垂直和单位点配位(图5d),可能是由于Cl–在Pd表面的竞争性吸附。
此外,理论计算表明,EG优先以平行和双位配位结构吸附在纯Pd表面,随着CO2的产生,更倾向于进行C-C裂解。这一发现与以往的理论研究相一致相反,随着Pd表面Cl–覆盖率的增加,EG倾向于采用垂直的单位点配位构型,这反过来又抑制了C-C的裂解,促进了EG的解吸。这些行为与前面提到的FTIR光谱结果一致(图5),表明Cl的存在改变了EG的吸附构型,从而抑制了C-C裂解途径,有助于抑制CO2。
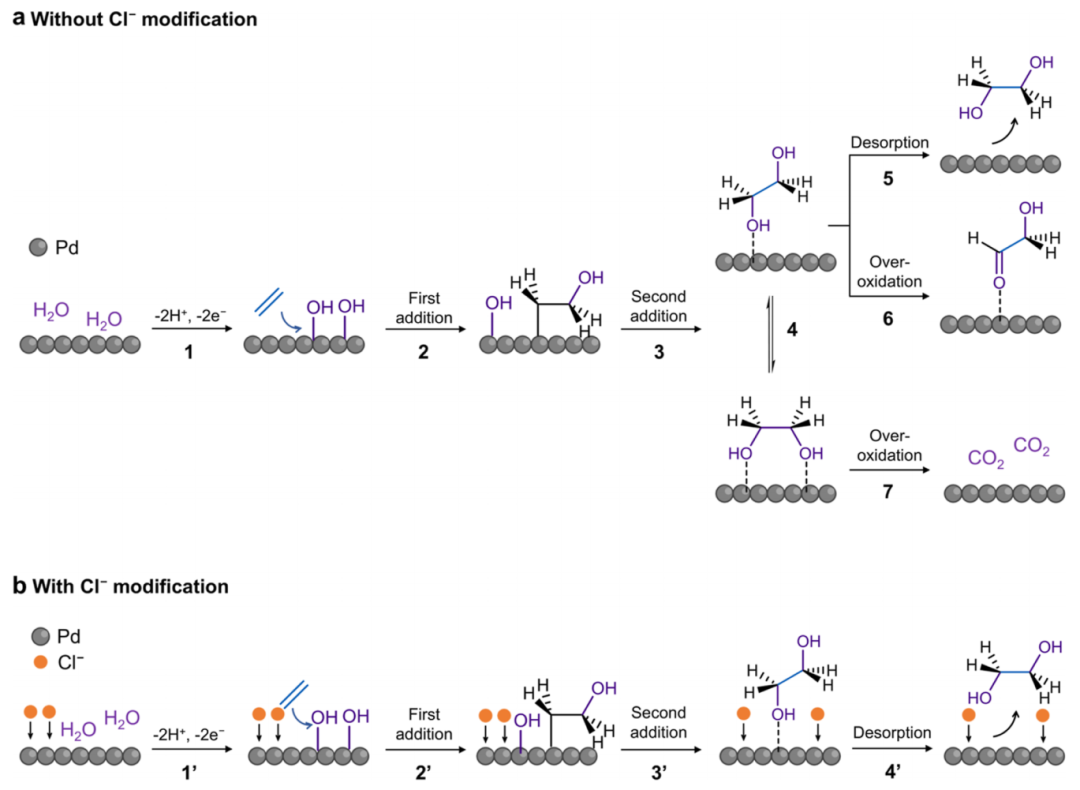
图6 C2H4氧化机理示意图
基于上述调查和先前的报道,提出了Pd ND和Pd ND-Cl的合理反应途径,并理解了促进Cl–的作用(图6)。在图6a中,水首先在Pd表面被活化,产生反应性的Pd-OH(步骤1)。然后,OH*物质依次加入到C2H4中(步骤2、3),最终生成EG。值得注意的是,在Pd表面现场生成的EG在两种吸附构型之间相互转换,包括单位点吸附和双位点吸附(步骤4)。因此,除了所需的EG解吸(步骤5)外,C2H4的过度氧化包括单位点吸附EG到GA(步骤6),双位点吸收EG到CO2(步骤7)和乙醇到AA/AcOH的途径同时发生,共同导致低的FEEG。
在Pd ND-Cl上(图6b), Cl–通过静电相互作用吸附在Pd表面。水活化和OH*连续加入C2H4(步骤1′-3′)的机理与Pd-ND相似。由于Cl–具有很强的吸电子能力,Pd-OH的生成(通过步骤1’)加快,但其氧化能力减弱。因此,促进了C2H4到EG的转化,同时抑制了EG的过度氧化,包括C2H4到GA和乙醇到AA/AcOH的途径。此外,由于Cl–的竞争性吸附,EG以垂直和单位点结构吸附,这反过来又阻止了C-C的裂解,从而完全抑制了CO2的产生。最终,EG以高选择性解吸发生(步骤4’)。
