

对于电催化研究人员而言,单原子催化剂(如CoN4)的精准建模是揭示氧还原(ORR)、析氢(HER)等反应机理的核心前提。本教程基于Materials Studio软件,提供一套高效、可复现的CoN4模型构建流程,专为吸附能与活性位点研究优化设计,助力从原子尺度解析催化本质。


教程核心价值:
1.靶向吸附研究:通过调控Co-N配位环境(键长1.8-2.0 Å)及基底电子结构,直接关联O₂、OH*等关键中间体的吸附强度,为火山图分析与过电位预测提供可靠模型。
2.实验-计算闭环:模型兼容实验表征数据(如XANES、EXAFS),可逆向指导合成中氮掺杂浓度与Co负载量的优化。
3.高效参数设置:涵盖石墨烯基底构建、氮空位精确刻蚀、Co原子配位优化等关键步骤,避免周期性边界干扰,确保吸附能计算精度。
典型应用场景:
· ORR反应:计算OOH与O与OH在CoN4位点的吸附自由能,定位决速步。
· CO₂还原:分析COOH*中间体的稳定化机制,筛选高效转化路径。
· 活性描述符提取:结合d带中心与Bader电荷,建立电子结构-活性关联模型。
无论您是探索新型单原子催化剂,还是深耕反应机理,均可通过此模型快速搭建理论-实验桥梁。
立即跟随教程构建您的CoN4模型,解锁电催化吸附研究的原子视角!
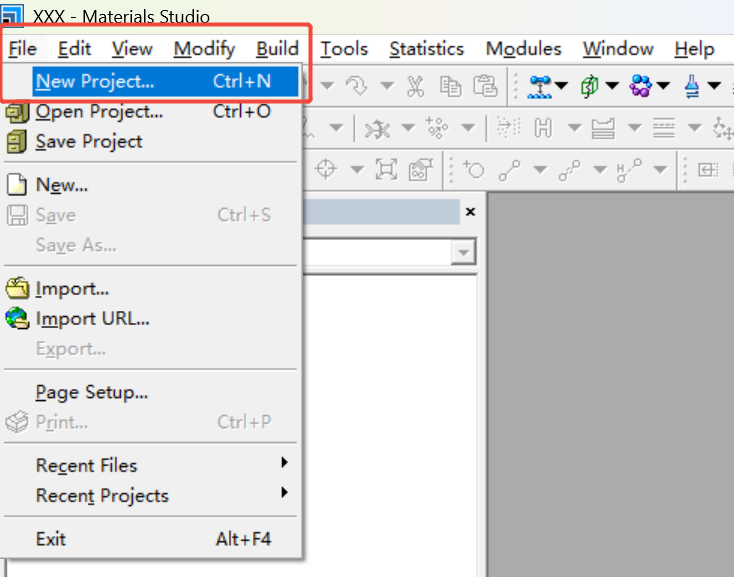
步骤1:创建石墨烯基底
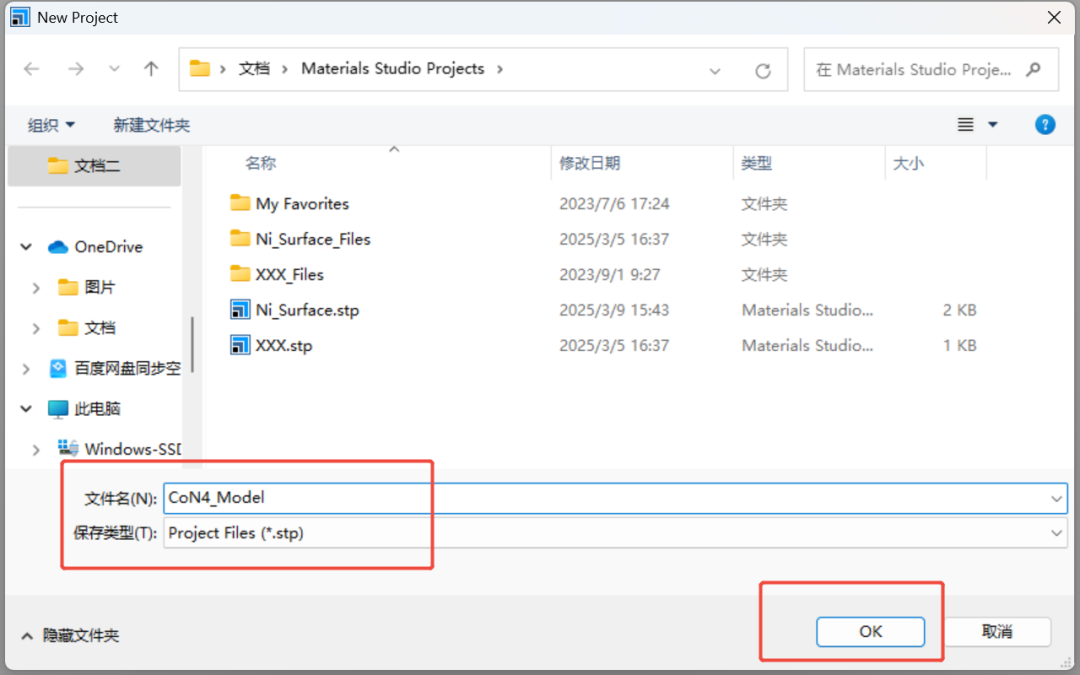
1.新建工程:打开MS → File → New Project → 命名(如“CoN4_Model”)
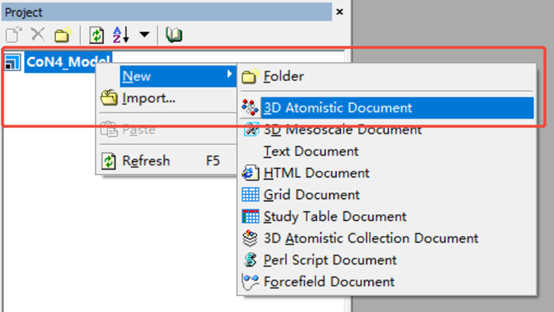
→选中左下角CoN4_Model点击鼠标右键→New→3D Atomistic.xsd
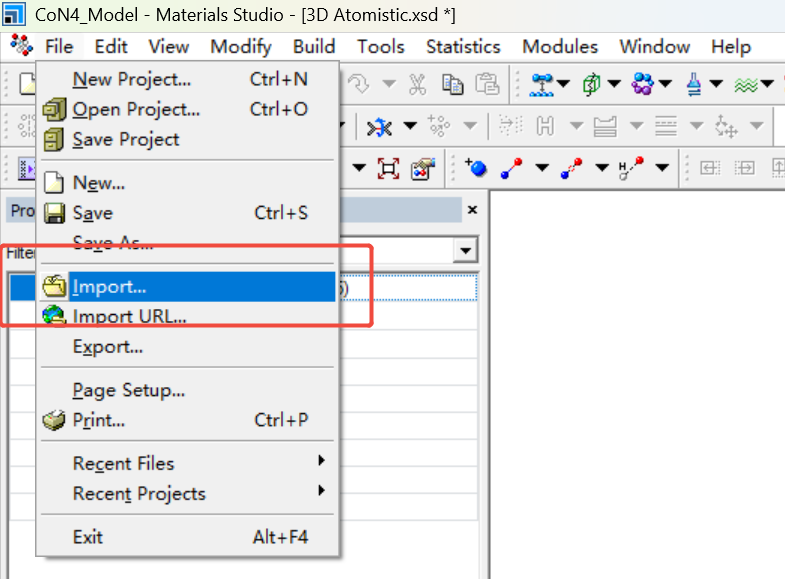
2.导入石墨烯:
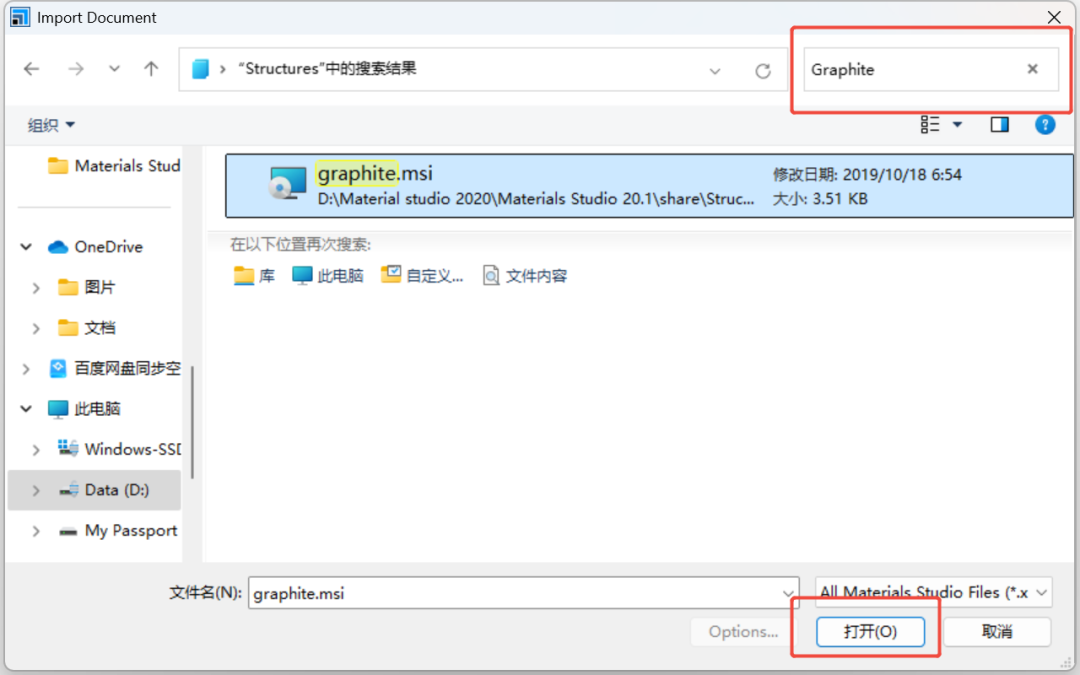
File → Import → Structures → 搜索Graphite(石墨结构)。
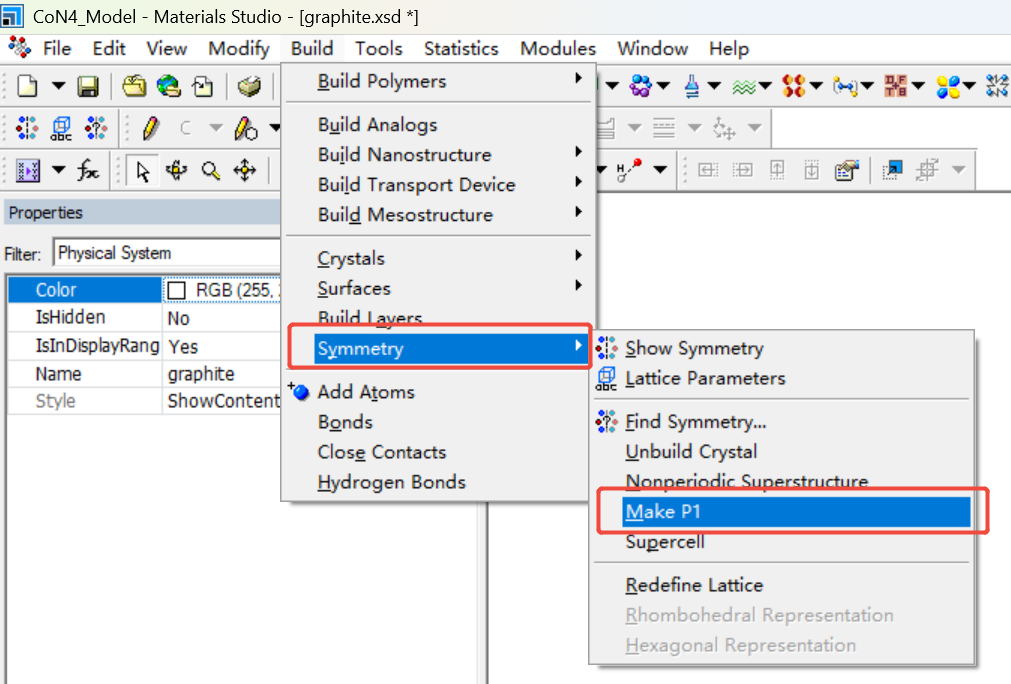
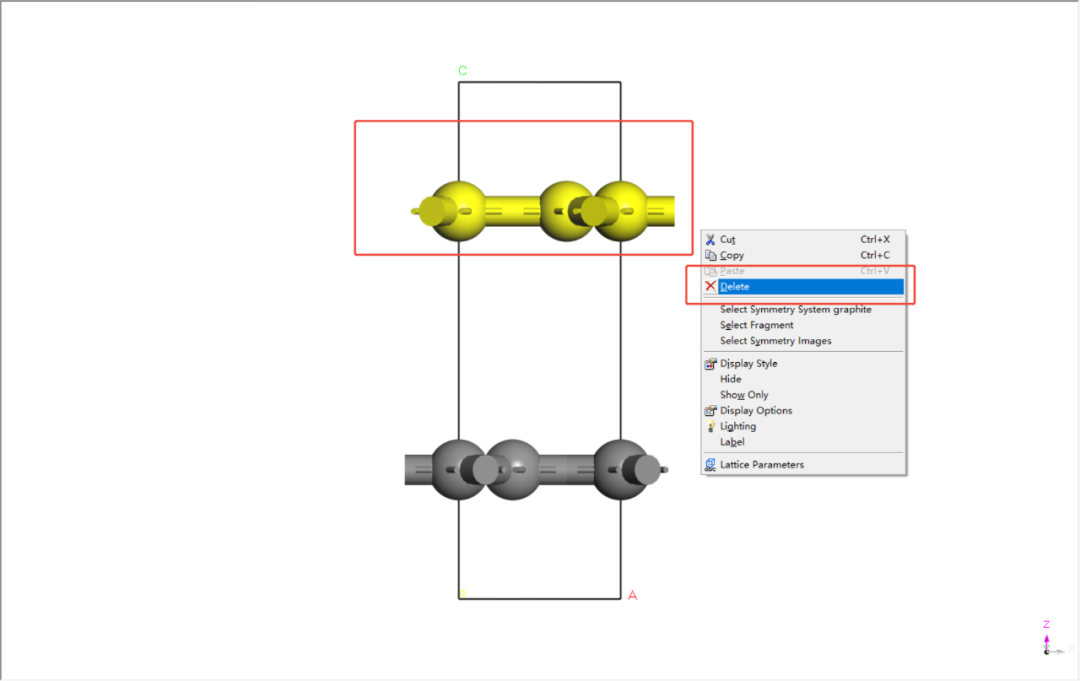
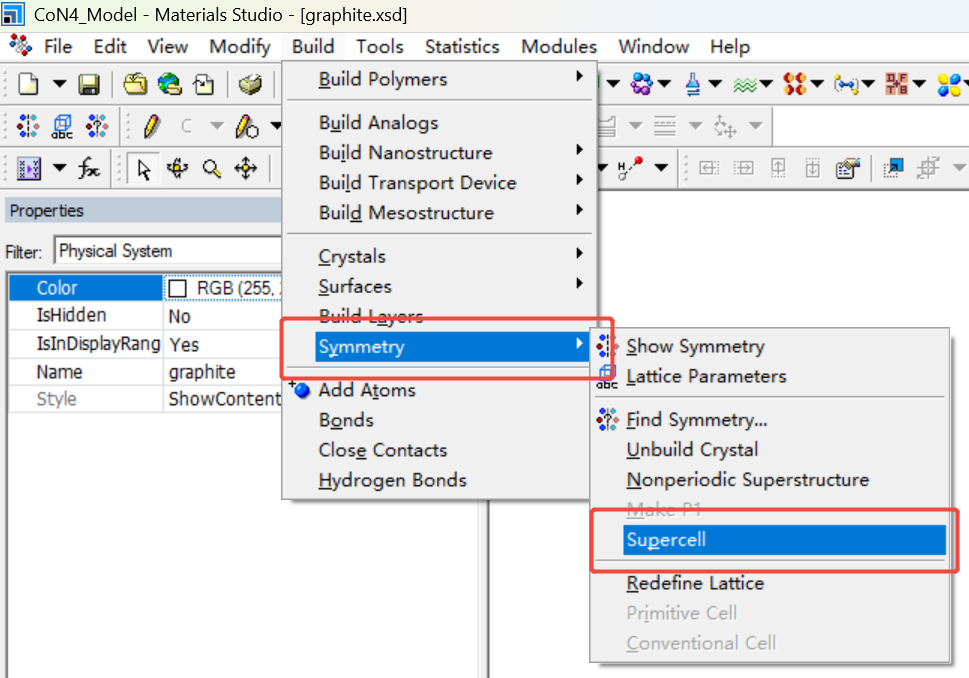
取消对称性,删掉一层碳原子并进行扩胞(6x6x1)处理。
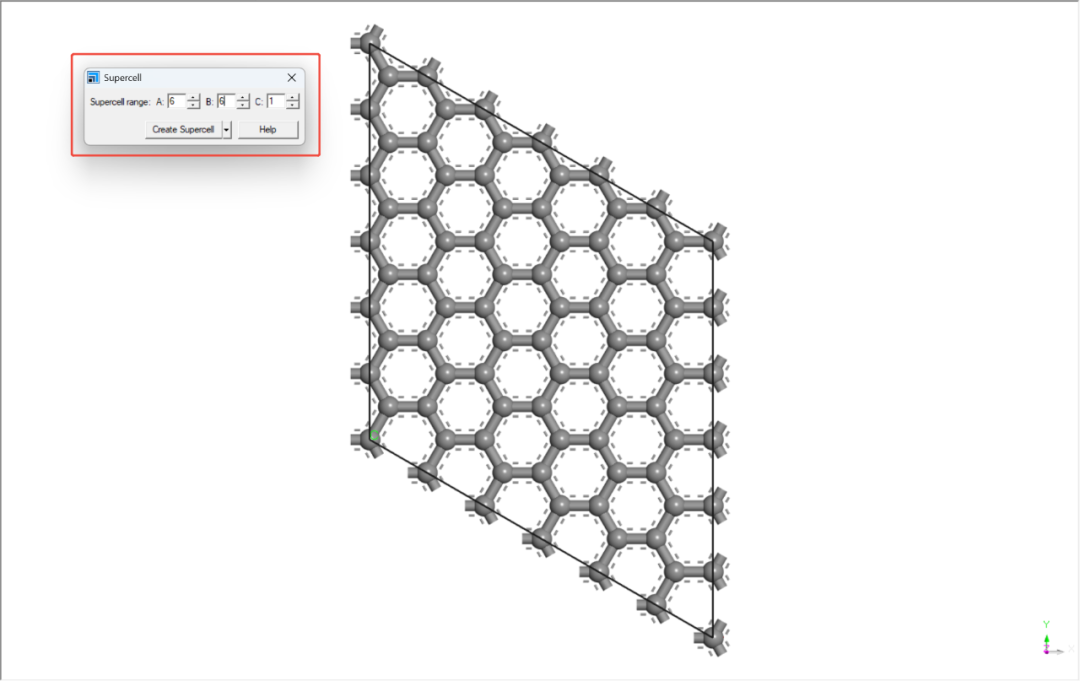
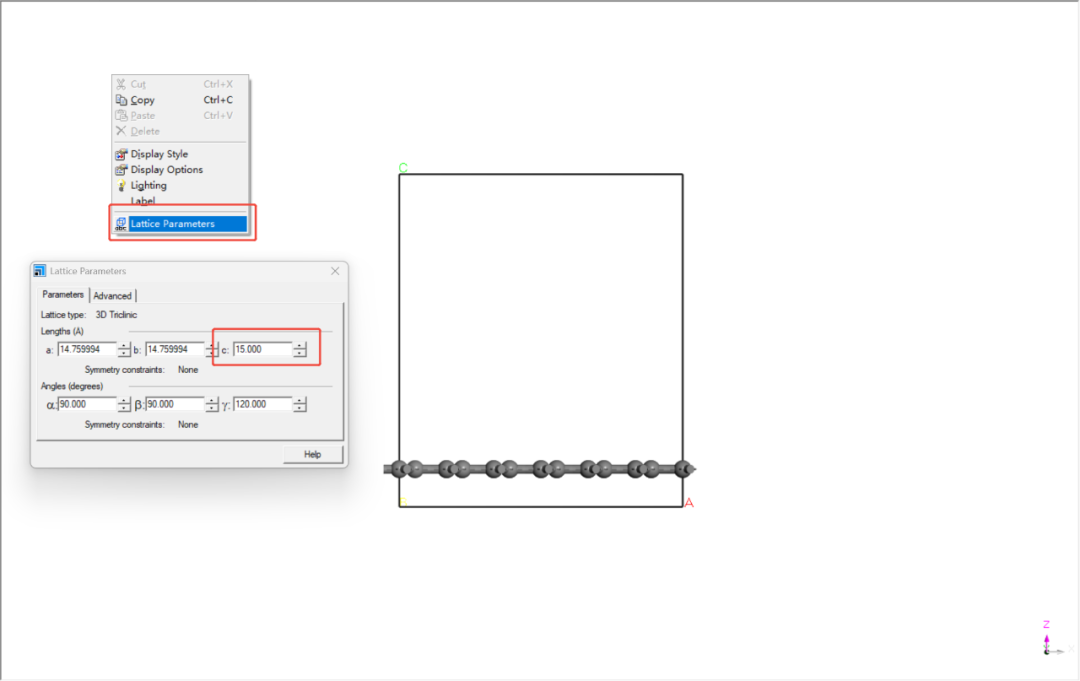
3.添加真空层:右键晶胞→ Lattice Parameters → 设置c轴真空层厚度≥15 Å。
注:改变c轴参数之前检查Advanced确保Keep fractional选项未被勾选。
步骤2:氮掺杂石墨烯基底
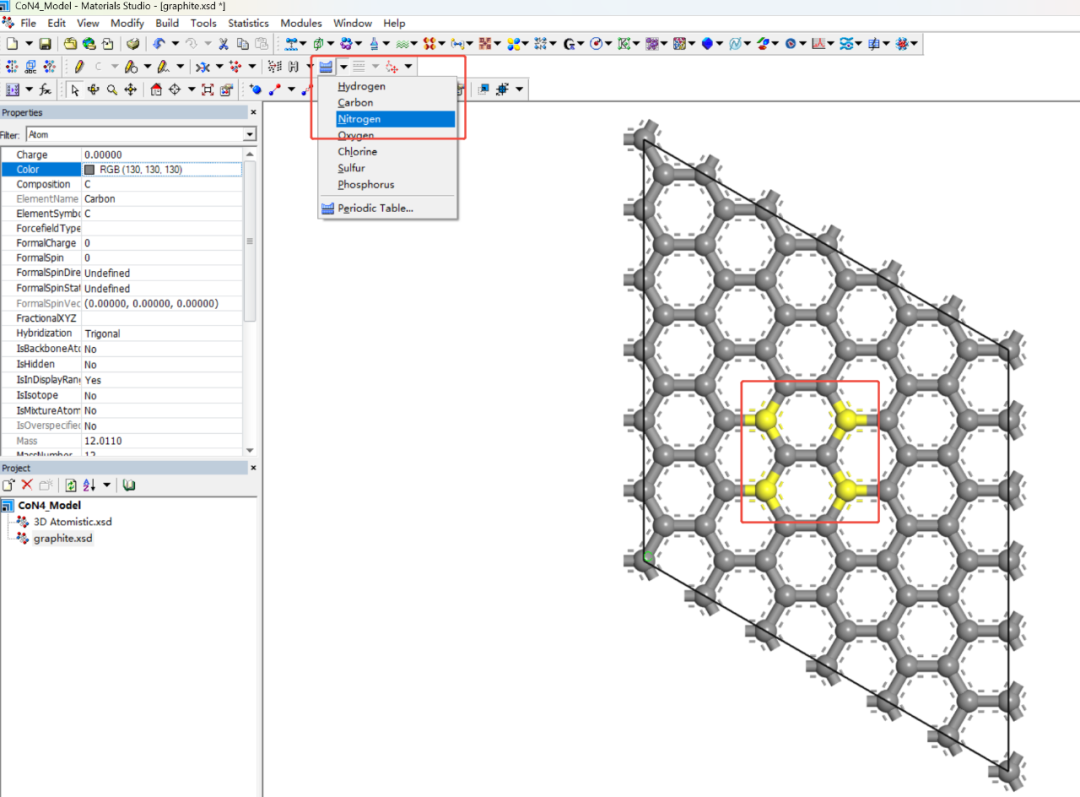
替换碳原子为氮:
– 选择石墨烯晶胞中4个对角的碳原子(形成正方形或菱形区域)。
– Shift+左键选中原子→ Modify → Element → 更改为N。
步骤3:创建CoN4配位空位
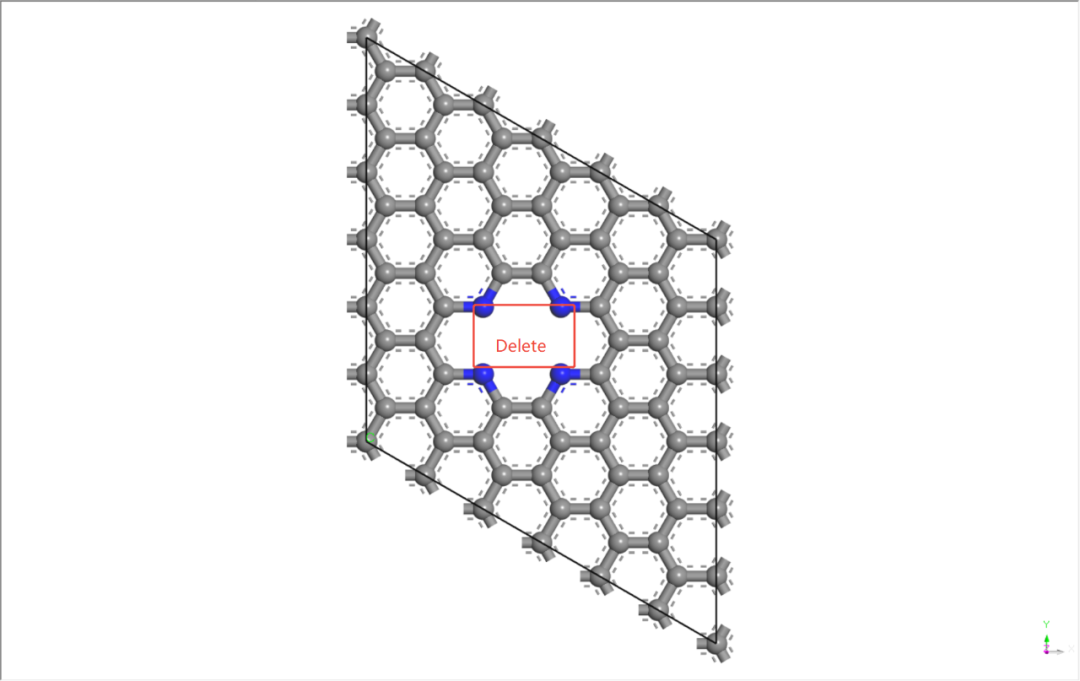
移除中心碳原子:
– 在4个N原子包围的中心位置选中2个C原子。
– Shift+左键选中原子 → Delete Atoms → 删除该C原子,形成空位。
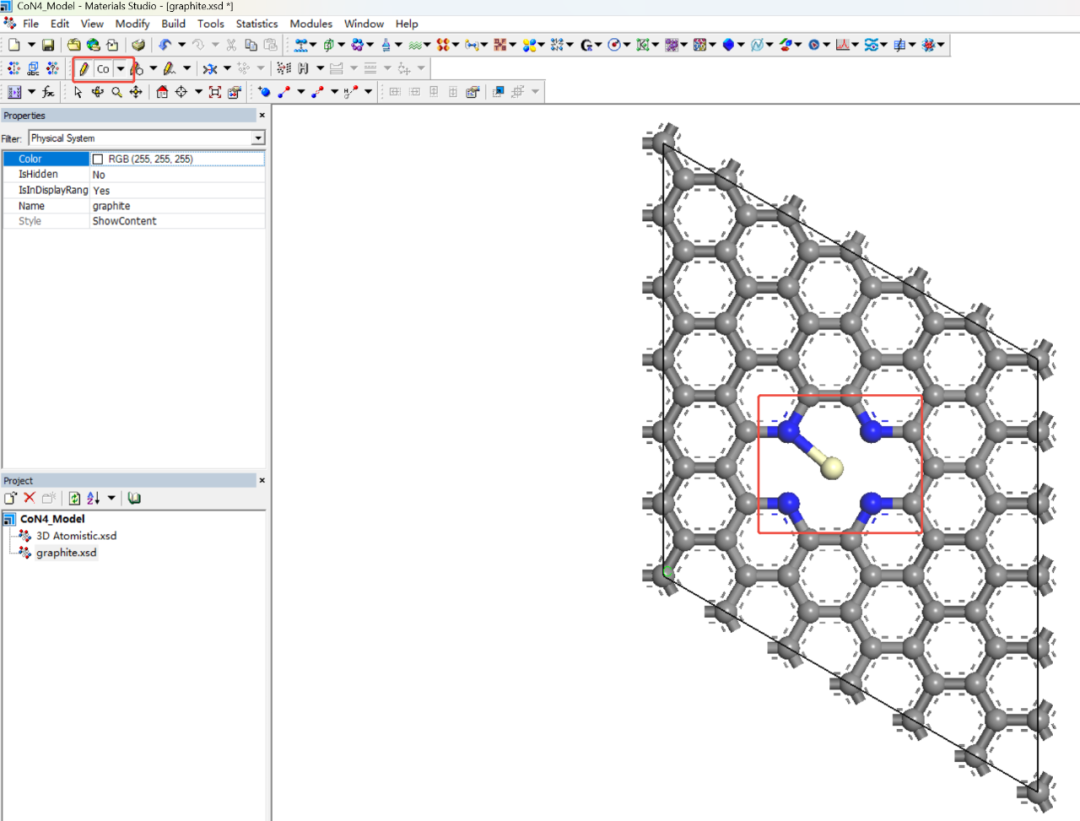
步骤4:添加Co原子形成CoN4结构
1.放置Co原子:
Sketch atom → 选择Co元素。
将Co原子精确放置在空位中心,与4个N原子形成配位。
2.调整键长:
手动拖动Co原子,确保Co-N键长在1.8-2.0 Å范围内(参考实验值)。
使用Measure/Change工具检查键长(Tools → Bond Length)。
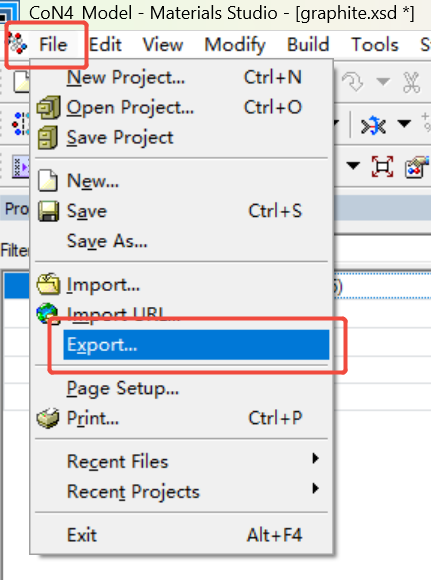
步骤5:导出成CIF文件
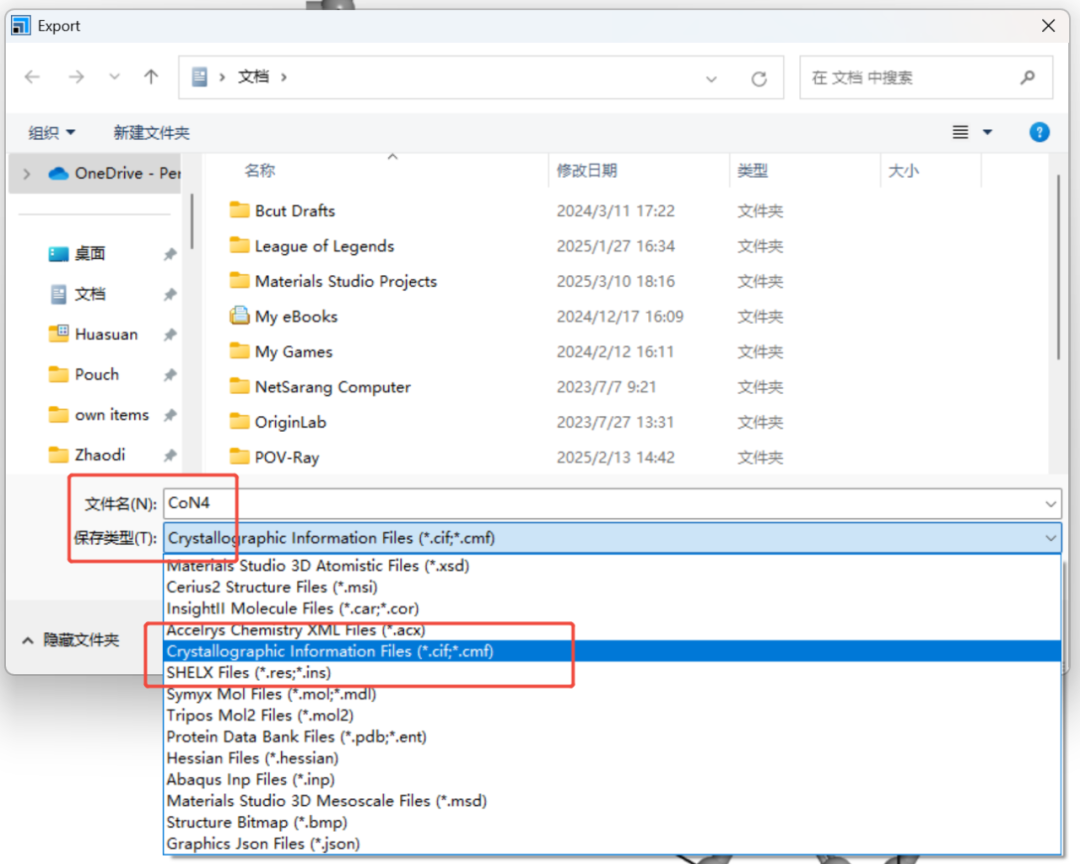
File→Export→选择CIF文件并命名为CoN4.cif进行保存即可。
通过以上步骤,您可以在Materials Studio中高效构建CoN4单原子催化模型。如需进一步电子结构计算(如DFT),可将优化后的结构通过VESTA另存为.vasp文件,供VASP或其他量子化学软件使用。
找华算做计算👍专业靠谱省心又省时!
益于理论计算化学的快速发展,计算模拟在纳米材料研究中的运用日益广泛而深入。科研领域已经逐步形成了“精准制备-理论模拟-先进表征”的研究模式,而正是这种实验和计算模拟的联合佐证,更加增添了论文的可靠性和严谨性,往往能够得到更广泛的认可。
华算科技已向国内外1000多家高校/科研单位提供了超过50000项理论计算和测试表征服务,部分计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。
