

近年来,基于甲酸的燃料电池备受关注,因其具有更高的开路电压和更低的交叉,可替代甲醇。当前纳米尺寸的Pt和Pd催化剂存在CO中毒和聚集问题,负载在氮掺杂碳上的单原子Rh或Ir以及单原子Pt合金催化剂被提出用于阳极的甲酸氧化反应(FAOR)。SACs因原子位点与配合物相似,具有超高的原子效率和质量活性,但其原子位点不可移动,难以同时与一个底物的多个位点结合。DAPs含有两个独立原子位点,可同时与一个底物的多个位点结合,调节底物与活性位点之间的电子相互作用,加速反应动力学,改善反应热力学。清华王定胜/中南熊禹/西交大黄正清团队开发了一种通用合成方法,获得DAP-(M, Rh)/CN(M = V, Cr, Mn, Fe, Co, Ni, Cu),其中含有相邻的原子Rh和M对。在DAP-(M, Rh)/CN中,单原子M位点对Rh位点在FAOR方面显示出巨大且不同的影响。Cr显示出最大的质量活性增强,达到64.1 A·mg⁻¹,比SA-Rh/CN(17.0 A·mg⁻¹)高3.8倍。随着M的原子序数的增加或减少,DAP-(M, Rh)/CN的质量活性降低。DAP-(V, Rh)/CN显示出大幅下降,仅为11.7 A·mg⁻¹,而DAP-(Ni, Rh)/CN和DAP-(Cu, Rh)/CN对FAOR几乎没有改善。DFT计算结合原位FTIR光谱表明,单原子M位点吸附HCOO物种,并与邻近的Rh位点协同作用,使得H原子以加速FAOR。该成果以“The Cooperative Effects of the Rh-M Dual-Metal Atomic Pairs in Formic Acid Oxidation”标题发表于《Angew. Chem. Int. Ed.》上。



图1. 结构表征。a) DAP-(M, Rh)/CN 的制备策略示意图。b) DAP-(Cr, Rh)/CN 的透射电镜(TEM) 图像。c) DAP-(Cr, Rh)/CN 的EDS)元素分布图,分别显示了碳 (红色)、氮 (蓝色)、铬 (黄色) 和铑 (绿色) 的分布。d) DAP-(Cr, Rh)/CN 的原子分辨率高角度环形暗场扫描透射电子显微镜(AC-HAADF-STEM) 图像和 e)放大图像,通过 Z 对比度分析,典型的铑和铬原子分别用黄色和蓝色在绿色矩形中圈出,M 和铑单原子分别用紫色和橙色圈出。f) DAP-(Cr, Rh)/CN 的铑 K 边扩展 X 射线吸收近边结构 (XANES) 和 g) 傅里叶变换扩展 X 射线吸收精细结构 (FT-EXAFS) 谱图,以及参考样品的谱图。h) SA-(Cr, Rh)/CN 的铬 K 边 XANES 和 i) FT-EXAFS 谱图,以及参考样品的谱图。
本研究采用ZIF-8的“主体–客体”策略原位捕获Rh和M金属前驱体,利用其大空腔和小孔径特性,在1000℃、6小时的高温下使Zn位点被蒸发并被Rh和M取代形成DAP-(M, Rh)/CN。以DAP-(Cr, Rh)/CN为例,其XRD图谱仅显示石墨碳的两个峰,SEM和TEM图像表明其保留了ZIF-8的菱形十二面体形貌且表面变得粗糙,EDS图谱显示C、N、Cr和Rh均匀分布,BET等温线和孔径分布显示其保留了ZIF-8的高比表面积和微孔体积,ICP-OES测得金属含量分别为0.70 wt.%(Cr)和0.67 wt.%(Rh)。ACHAADF-STEM图像显示Rh和Cr原子以原子对形式存在,原子间距为4.03 Å,且Rh-Cr对与Rh单原子的比例为69.1%。XAFS分析表明,Rh和Cr在DAP-(Cr, Rh)/CN中以原子形式分散,具有特定的配位结构和氧化态,且其XANES光谱与理论模型模拟结果相符,进一步的DFT计算采用相邻的Rh-N₄和Cr-N₄基团。
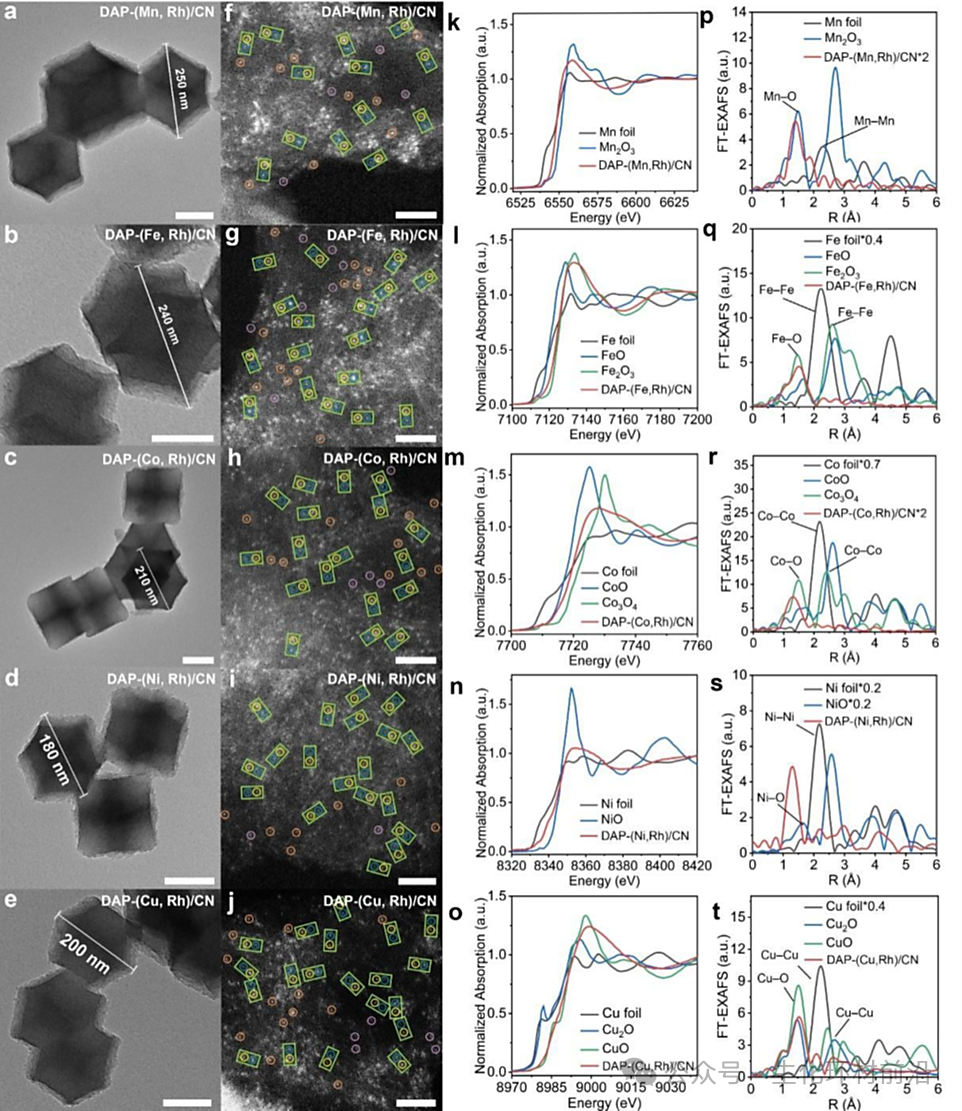
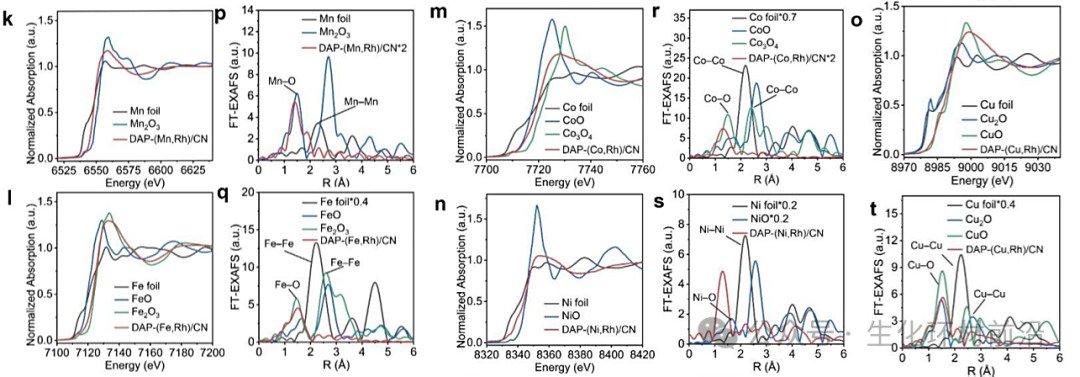
图 2. DAP-(M, Rh)/CN 的表征。a-e) 透射电镜(TEM) 图像。f-j)原子分辨率高角度环形暗场扫描透射电子显微镜(AC-HAADF-STEM) 图像,在绿色矩形中,典型的铑(Rh) 和 M 原子分别用黄色和蓝色圈出,而 M 和铑单原子则分别用紫色和橙色圈出。k-o) DAP-(M, Rh)/CN 的扩展 X 射线吸收近边结构 (XANES) 谱图和 p-t) 傅里叶变换扩展 X 射线吸收精细结构 (FT-EXAFS) 谱图。
为验证合成策略的普适性,制备并表征了一系列 DAP-(M, Rh)/CN(M = Mn、Fe、Co、Ni、Cu),其形貌均为边长约200 nm 的菱形十二面体,未检测到团簇或纳米颗粒。ICP-OES 测得 M 和 Rh 的负载量分别为 0.37-1.38 wt.% 和 0.67-0.95 wt.%。其比表面积和孔径分布与DAP-(Cr, Rh)/CN 相似。从 ACHA-ADF-STEM 图像中可观察到大量相邻的 M-Rh 原子对,原子间距约为 3-5 Å,且 Rh-M 对是主要组分。XPS表明DAP-(M, Rh)/CN 中的 C 和 N 物种与 DAP-(Cr, Rh)/CN 中的无明显差异,M 物种带正电。DAP-(M, Rh)/CN 在 Rh K 边XANES 光谱中的结构与 DAP-(Cr, Rh)/CN 中的相似,且 Rh和 M 位点呈原子分散状态。
利用 XAFS 拟合曲线及相关参数识别 Rh 和 M 的配位结构,结果表明存在Rh-N4 基团且轴向吸附有两个 O,M 位点具有类似的 M-N4 结构。为探究双原子对催化剂的形成机制,进行了热解实验,发现 Rh单原子并非直接来源于前驱体,而是纳米颗粒的再分散。合成具有不同 Cr 负载量的 DAP-(Cr, Rh)/CN,发现高剂量的Cr(acac)3 导致 Cr 原子团聚,且过量的 M 原子有助于双原子对的形成。根据实验结果和之前的研究,采用催化剂模型并计算能量变化,发现异核DAP-(V, Rh)/CN 和 DAP-(Cr, Rh)/CN 等具有较高的稳定性,表明高温热解提高金属原子流动性并克服能量障碍,结合 Zn蒸发留下的活性位点,是实现双原子对催化剂的关键因素。
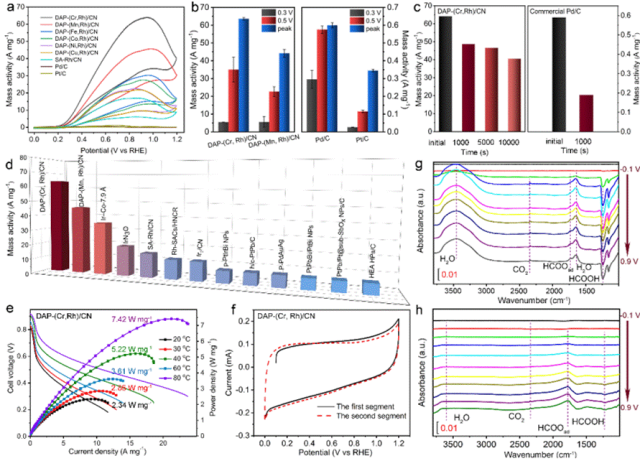
图3. 甲酸电氧化性能。a) DAP-(Cr, Rh)/CN、DAP-(Mn, Rh)/CN、Pd/C和Pt/C 在含甲酸(0.5 M) 的硫酸水溶液(0.5 M) 中的循环伏安(CV) 曲线,扫描速率为 10 mV s−1,测试电位分别为0.3 V、0.5 V 和峰值电位。b) 上述催化剂的质量活性。c) DAP-(Cr, Rh)/CN 和Pd/C 在加速老化测试(CA) 前后的质量活性。d) DAP-(Cr, Rh)/CN、DAP-(Mn, Rh)/CN 和近期报道的催化剂的质量活性对比。e) 基于 DAP-(Cr, Rh)/CN 的直接甲酸燃料电池(DFAFCs) 在不同温度下,以 3 M 甲酸为燃料的稳态极化曲线和功率密度曲线,甲酸流速为 1 mL/min,氧气流速为750 mL/min,无背压。f) DAP-(Cr, Rh)/CN 的一氧化碳(CO) 剥离测试。g) h) DAP-(Cr, Rh)/CN 和 g) SA-Rh/CN 的原位傅里叶变换红外光谱(FTIR)。
为了探究 FAOR 中相邻 M 位点的协同效应,评估了 DAP-(Cr, Rh)/CN 等催化剂的性能。结果表明,DAP-(Mn, Rh)/CN 在峰值电位时电流密度最高,是商业Pd/C 的 3.7 倍;DAP-(Cr, Rh)/CN 的质量活性在峰值电位时达 64.1 A·mg⁻¹,显著高于其他催化剂。其在不同区域的质量活性均远超Pd/C 和Pt/CN 。不同 Cr 负载量且Cr-Rh 对比例低的情况对 FAOR 活性较低,证实Cr-Rh 原子对提升性能关键。DAP-(Cr, Rh)/CN 稳定性好,计时电流法测试后质量活性仅损失25%,加速稳定性测试5000 次循环后仍保持87% 性能,且其在DFAFCs 中应用潜力大,相关 DFAFCs 性能优异。系列测试揭示反应机理,CO 剥离测试及动力学同位素效应表明C-H 键断裂为速控步,反应主要通过甲酸盐路径进行。
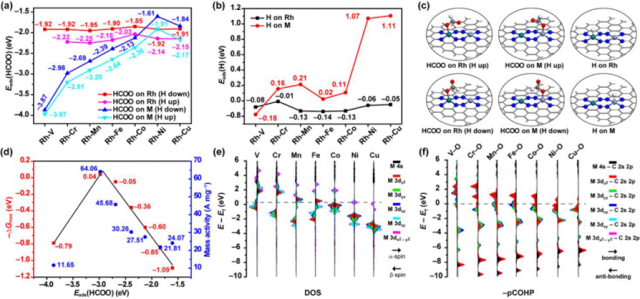
图4. 通过密度泛函理论(DFT)模拟对反应机理的研究。a, b) 关键中间体HCOO(a)和H(b)在Rh位点和M位点上的吸附能。H向上和H向下的符号分别表示HCOO的H原子远离金属位点和靠近金属位点的吸附构型。c) HCOO和H在Rh位点和M位点上的吸附结构。d) 通过甲酸盐路径在FAOR(甲酸氧化反应)中最大吉布斯自由能的负值(–ΔGmax)以及DAP-(M, Rh)/CN催化剂的质量活性,二者均作为过渡金属位点上HCOO吸附能的函数。e) 清洁Rh-M位点中M原子的投影态密度。f) 吸附有HCOO物种的Rh-M位点中M–O键的负pCOHP曲线。
为了探究 M 和 Rh 原子在 FAOR 中的协同效应及反应机理,通过理论计算与实验验证相结合的方法,取得了以下重要发现:理论计算表明,HCOO在 Rh-M 位点的 M 原子上的吸附能从 V 到 Ni呈线性下降,后略有上升,这与 FAOR 活性趋势相符,且 Rh原子更倾向于吸附 H 原子,而 M 原子则稳定反应中间体。实验合成的DAP-(V, Rh)/CN 催化性能较低,验证了理论预测的准确性。进一步的吉布斯自由能计算和电子结构分析显示,Rh-Cr 和 Rh-Mn 位点在甲酸盐路径上具有热力学优势,且随着 M 原子价电子增加,HCOO吸附强度减弱,脱氢能量提高,揭示了 Rh和 M 位点协同效应的电子机制。
在这项工作中,该团队开发了 DAP-(M, Rh)/CN(M = V, Cr, Mn, Fe, Co, Ni, Cu)的一般合成方法。DAP-(M, Rh)/CN 中 M 的变化导致 FAOR 的催化性能有了显著不同的提升。DAP-(Cr, Rh)/CN 展现出最佳的 FAOR 活性,其最高质量活性为 64.1 A·mg⁻¹。随着 M 的原子序数增加,M 和 Rh 金属原子之间的协同效应逐渐减弱。在原位 FTIR 光谱中直接检测到了关键中间体(HCOOad)。通过 DFT 计算揭示了反应机理以及 Rh和 M 单原子位点之间的协同效应:在一系列 DAP-(M, Rh)/CN催化剂上,甲酸通过甲酸盐路径氧化,其中辅助 M 金属结合 HCOO 物种,Rh 原子接受 H 原子,这减轻了位阻并促进了反应动力学。此外,HCOO在 Cr位点上最适宜的吸附能有助于两个自发的基元反应步骤,因此DAP-(Cr, Rh)/CN 在FAOR 中表现出最高的质量活性。相信这项工作为未来FAOR 催化剂的开发指明了一条新途径。
原文链接:
10.1002/anie.202503095
测试目的
✅探究催化剂结构:通过X射线吸收光谱(XAS)技术,研究负载在掺氮碳催化剂上的Rh-M双金属原子对(DAP-(M, Rh)/CN,M = V, Cr, Mn, Fe, Co, Ni, Cu)的结构特性,包括金属原子的配位环境、价态以及原子间的距离等,以深入了解催化剂的活性中心结构。
✅揭示协同作用机制:分析不同M原子与Rh原子组成的双原子对在催化反应中的相互作用,探究M原子对Rh原子电子结构和催化性能的影响,从而揭示双原子对协同促进甲酸氧化反应(FAOR)的机制。
测试原理
✅X射线吸收边近边结构(XANES):当X射线照射到样品时,入射光子能量与原子内电子能级跃迁相对应,不同能量的光子被吸收,形成X射线吸收边。XANES谱主要反映吸收边附近的信息,可用于确定金属原子的价态、电子结构以及对称性等。通过比较不同样品在相同吸收边处的XANES谱,可以分析金属原子的氧化态和电子态的变化。
✅扩展X射线吸收精细结构(EXAFS):在吸收边以外的区域,X射线与原子内电子相互作用产生干涉效应,形成精细结构。EXAFS谱主要反映吸收原子周围邻近原子的信息,包括原子间的距离、配位数以及原子种类等。
测试结果
✅判断价态:以DAP-(Cr, Rh)/CN为例,吸收边能量接近,说明Rh在DAP-(Cr, Rh)/CN中的氧化态为+3。DAP-(Cr, Rh)/CN的Cr K-edge XANES谱吸收边能量位于Cr箔和Cr₂O₃之间,确认Cr位点带正电。
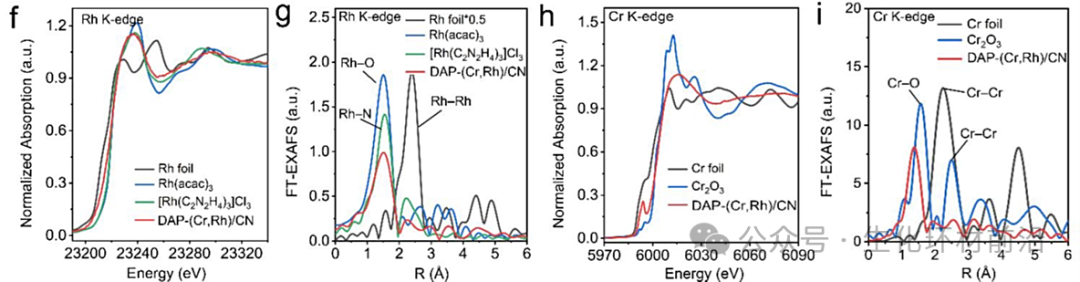
✅判断配位环境:FT-EXAFS谱在1.50 Å处出现一个主峰,介于[Rh(C₂N₂H₄)₃]Cl₃的Rh-N峰和Rh(acac)₃的Rh-O峰之间,表明DAP-(Cr, Rh)/CN中同时存在Rh-N和Rh-O键。同理FT-EXAFS谱在1.37 Å处出现一个主峰,归属于Cr-N(O)散射路径,表明Cr原子在DAP-(Cr, Rh)/CN中以单原子形式分散并与N(O)配位。
对于其他DAP-(M, Rh)/CN(M = Mn, Fe, Co, Ni, Cu),M K-edge XANES谱和XPS M 3d谱显示M物种在DAP-(M, Rh)/CN中带正电,且FT-EXAFS谱仅观察到M-N(O)峰,表明M原子也以单原子形式分散并与N(O)配位。
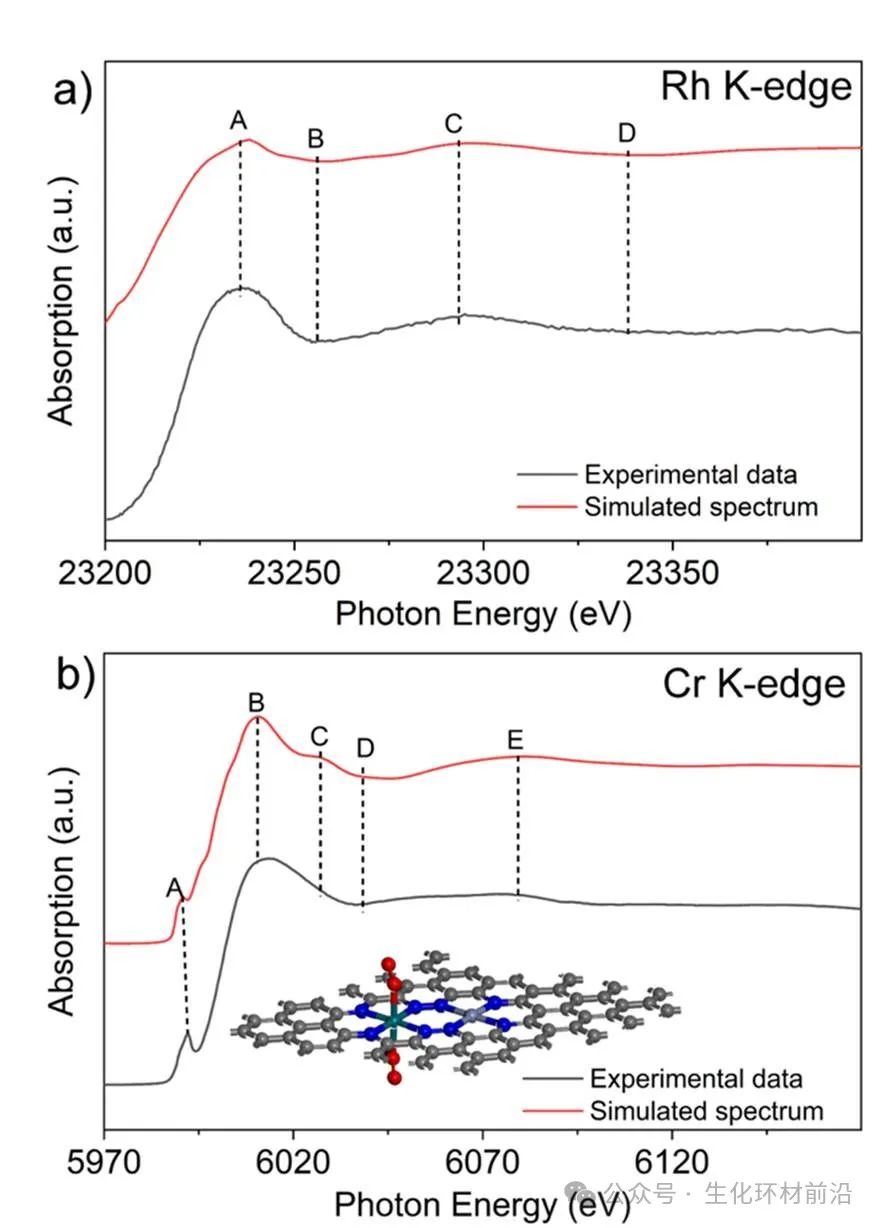
✅判断构型:Rh K-edge XANES谱图形状与Rh(acac)₃和[Rh(C₂N₂H₄)₃]Cl₃相似,表明Rh具有类似的六配位八面体构型,通过对 Rh 和 Cr K 边的 XANES 谱进行模拟,实验与模拟的 Rh K 边 XANES 谱具有相似特征。另外,模拟的 XANES 谱在 Cr K 边的前边峰 A 和后边峰 B 到 E 处与实验 XANES 谱表现出相似特征,即相邻的Rh-N₄和Cr-N₄基团。
M K-edge XANES光谱显示,DAP-(M, Rh)/CN中的M呈正电荷。在DAP-(M, Rh)/CN的Rh K-edge XANES光谱中表征的结构与DAP-(M, Rh)/CN相似,表明其具有相同的配位结构。在Rh和M K-edge的FT-EXAFS光谱中,只能观察到唯一的Rh-N(O)和M-N(O)峰,说明DAP-(M, Rh)/CN中Rh和M位点是原子分散的。

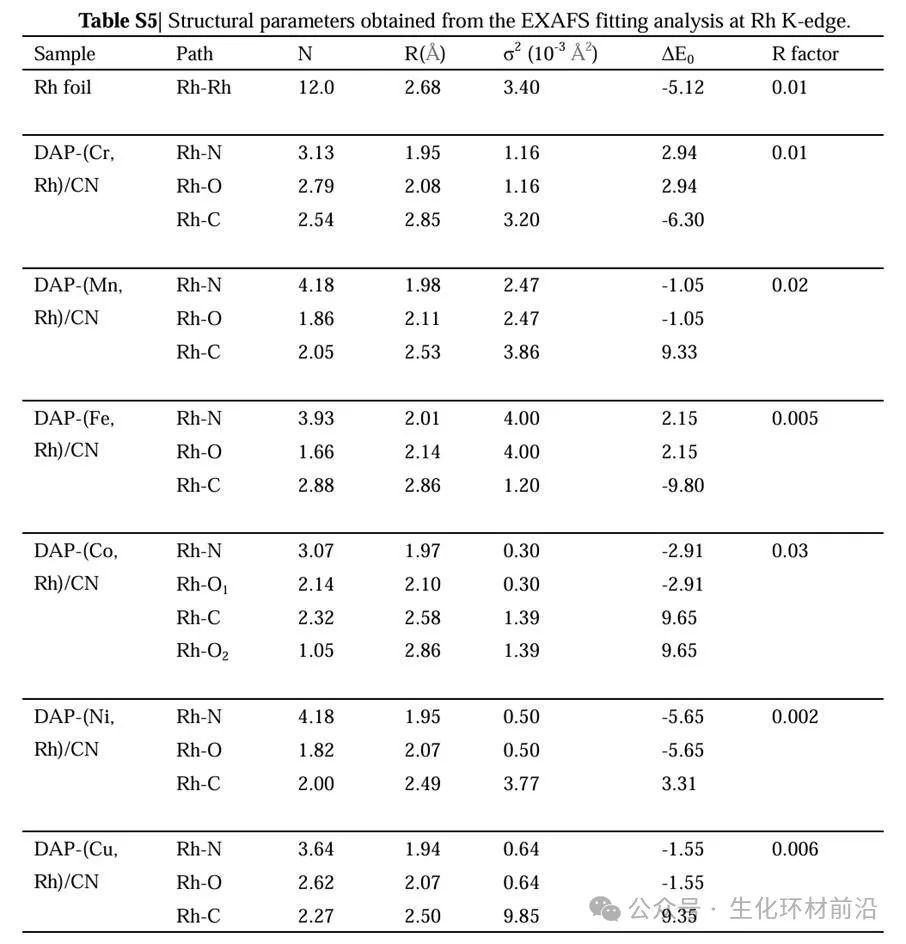
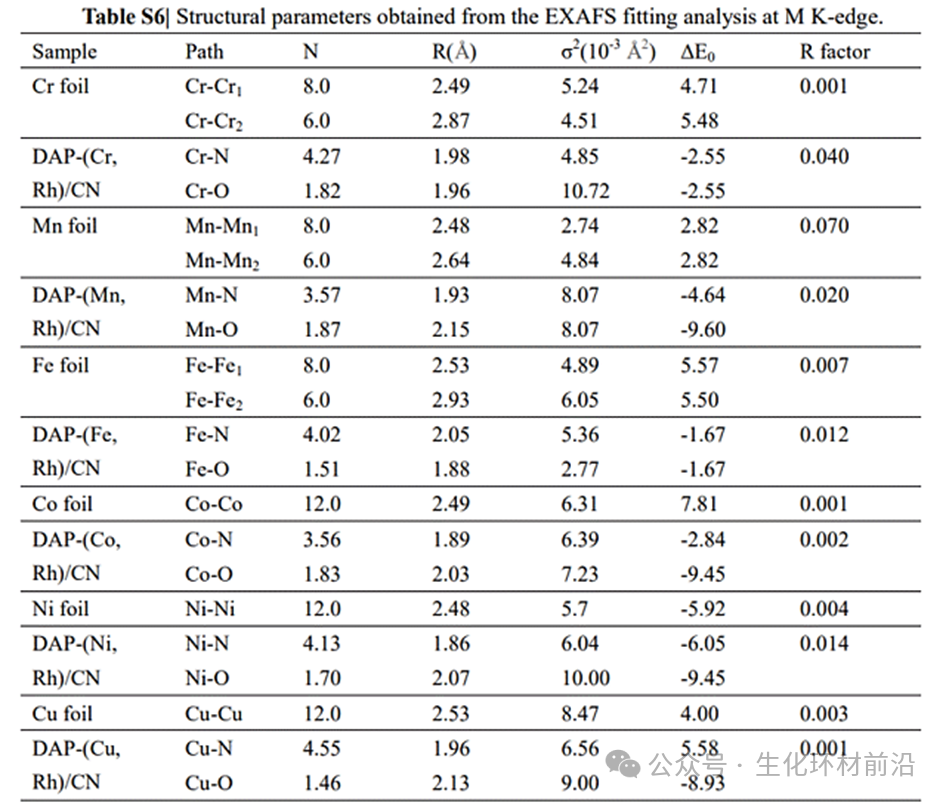
✅配位数及键长:XAFS拟合曲线和定量参数进一步确定了Rh和M的配位结构,表明Rh和M原子在DAP-(M, Rh)/CN中均具有类似的配位环境,如Rh-N和M-N的配位数约为4.0,键长分别约为1.95 Å和相应的长度,说明Rh和M原子在催化剂中形成了稳定的配位结构。

王定胜
王定胜,2000.09-2004.07在中国科学技术大学化学物理系攻读并获得学士学位,2004.09-2009.07于清华大学化学系攻读博士学位。2009.07-2012.06在清华大学物理系从事博士后研究工作,2012.07-2012.12担任清华大学化学系讲师,2012.12-2023.12晋升为清华大学化学系副教授,2023.12至今担任清华大学化学系教授。其研究领域主要是金属纳米晶、团簇及单原子为主的无机功能纳米材料的合成、结构调控与催化性能。在Nature、Nature Catal.、Nature Chem.、Nature Nanotech.、Angew. Chem. Int. Ed.、J. Am. Chem. Soc.等学术期刊发表200余篇论文,2012年获全国百篇优秀博士论文奖,2013年获国家优秀青年基金,2018年获青年拔尖,2023年获国家杰出青年基金,2020-2023年连续四年入选全球高被引科学家。
熊禹
熊禹,2009.09-2013.06在大连理工大学应用化学系攻读并获得学士学位,2013.09-2018.05于中南大学攻读化学理学博士学位。2018.07-2020.07在清华大学从事博士后研究工作,2020.10至今担任中南大学化学化工学院特聘副教授。其研究领域为单原子,纳米催化剂合成及其性能研究。近年来,在国际期刊发表SCI论文20余篇,其中以(共同)第一/通讯作者身份在Nature Nanotechnology、Advanced Materials、等化学类顶级期刊中发表论文15篇,发明专利1项。
黄正清
黄正清,2016.9-2020.7在西安交通大学攻读并获得博士学位。2020.9-2022.6担任西安交通大学助理教授,2022.7-2023.9担任西安交通大学助理教授、硕士生导师,2023.10至今担任西安交通大学助理教授、博士生导师。其研究领域包括(近)真实条件下催化反应机理的动态模拟,机器学习辅助催化新材料的理性设计,甲烷、氨、二氧化碳、氢等小分子活化与转化的理论计算研究。在Nature Nanotechnology,Nature Catalysis,Nature Communications,Energy & Environmental Science,ACS Catalysis,Journal of Catalysis等学术期刊发表SCI论文30余篇,论文总引用次数4758余次,H-因子为20。主持国家重点研发计划子课题(2024-2028)、国家自然科学基金–青年基金(2022-2024)、中国博士后基金面上(2022)等科研项目。
