



第一作者:刘人凤,涂文静
通讯作者:陈光需,赵云,裴安,贾艳艳
通讯单位:华南理工大学,华东理工大学
论文DOI:10.1021/jacs.4c17193
利用可再生能源进行的低能耗电催化氧化苯甲醇制苯甲酸具有重要意义。为了解决目前电催化氧化制苯甲酸过程中存在的高电压输入(高能耗)、副反应和催化剂失活等问题,本研究提出了一种具有PtZn金属间结构的PtZn-ZnOx催化剂,其表面具有丰富的PtZn-ZnOx界面,可以在0.2 V(vs RHE)的低电压下驱动苯甲醇氧化反应的发生,并在0.725 V(vs RHE)的低电压下实现高苯甲酸选择性(99.5%),比大多数报道的研究低0.6 V。与商业Pt/C相比,PtZn-ZnOx催化剂的苯甲醇氧化起始电位降低了160 mV,在相同电位下,电流密度是商业Pt/C的两倍。为了深入揭示PtZn-ZnOx催化剂上苯甲醇氧化及C-H键活化机制,本研究通过结合实验和密度泛函理论计算,证明了PtZn-ZnOx界面中的不饱和配位Zn原子促进了苯甲醇和苯甲醛的吸附和亲电OH*的生成,且PtZn-ZnOx界面降低了OH*与苯甲醛偶联的能垒,从而提高了催化活性和选择性。此外,PtZn-ZnOx在低电位下对多种醇类有机小分子(如脂肪醇、芳香醇、糠醇等)表现出良好的电氧化活性,证明了本研究所述工程界面策略的普适性,有望为开发高效的电催化氧化醇类有机小分子催化剂提供新的思路。
可再生能源驱动的生物质增值在实现能源储存和可再生精细化学品的生产中起着至关重要的作用。生物质衍生苯甲醇(Ph-CH2OH)因其能有效转化为高值苯甲酸(Ph-COOH)而具有重要意义。苯甲酸是一种珍贵的增值化学品,在合成纤维、树脂和防腐工业中越来越多地用作必需的化学中间体。目前,传统工业生产苯甲酸主要依靠甲苯氧化工艺,其反应条件苛刻,通常需要高温(140 ~ 160℃)、高压(0.2 ~ 0.3 MPa)、化学添加剂(如KMnO4、酸性溶剂、溴化物促进剂)的加入,并伴有复杂的提纯过程(蒸馏、重结晶)。这一过程涉及到较高的碳排放。而本研究所报导的常温下以水为氧源的电催化苯甲醇氧化反应(BAOR)制备苯甲酸方法与目前广泛研究的好氧氧化和光催化氧化方法相比,具有反应条件温和、产物纯度高、分离纯化过程简化、催化剂回收方便等优点,近年来成为生产苯甲酸的重要替代方法。此外,只有两个稳定氧化产物的电催化BAOR能够作为简化涉及多个产物和中间体的复杂反应体系的理想探针和模型系统。由于C-H键活化是许多有机小分子氧化反应中的基本模块和关键过程,这种简化有助于升级生物质原料(如醇、糠醛、甘油)的研究,使C-H键活化的详细机制研究成为可能。
目前,电催化BAOR的通常需要高氧化电位,这导致高电压输入和能量消耗。为了提高苯甲醇选择性电氧化制备苯甲酸的效率,研究人员在电极材料的开发中做了大量的努力,其中过渡金属氧化物和氢氧化物(如Ni和Co氧/氢氧化物)被广泛研究,。在这些氧/氢氧化物催化剂上的电催化BAOR通常遵循亲核氧化反应机制。这种机制需要高氧化电位来生成活性金属(高价态),然后这些活性金属被亲核试剂(苯甲醇)还原回较低的氧化态。可惜的是,在高电压下工作会导致能源消耗增加和竞争性副反应的发生,如OER或电极材料的溶解/衰退。因此,开发具有高活性(高电流密度)、稳定性、低电压输入()和高苯甲酸选择性的BAOR先进催化剂势在必行。
先前的研究表明,贵金属基催化剂可以显著降低醇氧化所需的电位。例如,清华大学段昊泓团队在CoOOH上负载Au可以将BAOR所需的起始电位降低到0.6 V(vs RHE)。据报道,Pt基催化剂在BAOR中可以达到低至0.4 V (vs RHE)的起始电位,但在低电位下工作面临低BAOR电流密度和低苯甲酸选择性的问题。对于Pt基催化剂而言,有研究人员为了提高Pt催化剂在醇类有机小分子氧化中的催化性能,提出了双金属合金化工程。例如,金属间PtZn催化剂是甲醇或乙醇氧化的优良电催化剂,有研究人员利用理论计算证明了PtZn比原始Pt性能更好的原因是Zn原子对OH*中间体的稳定作用。此外,将Pt与Zn合金化构建PtZn催化剂不仅可以调整Pt的电子结构和d带中心,还可以促进相邻Zn原子产生孤立的Pt位点,从而通过减少醇氧化过程中的CO中毒来减缓催化剂的失活。研究表明,不饱和配位原子的表面边缘位点有助于催化反应,因此,结合Pt合金催化剂与表面不饱和配位原子的各自优势,有望开发在低电位下实现电流密度大、苯甲酸选择性高且稳定性强的BAOR催化剂。
(1) 本研究制备的PtZn-ZnOx催化剂可以实现在0.725 V(vs RHE)的低电压(比大多数报道的研究低0.6 V)下进行高效的苯甲醇氧化反应,并成功实现99.5%的苯甲酸选择性;
(2) 本研究通过结合实验和密度泛函理论计算证明了:PtZn-ZnOx界面中的不饱和配位Zn原子促进了苯甲醇和苯甲醛的吸附和亲电OH*的生成。此外,由于PtZn-ZnOx界面降低了OH*与苯甲醛偶联的能垒,从而提高了催化活性和选择性。
(3) PtZn-ZnOx在低电位下对多种醇类有机小分子表现出良好的电催化氧化活性,证明了本研究工程界面策略的普遍性。
本研究通过结合浸渍法和H2还原策略,成功制备了以PtZn金属间化合物(IMA)为主体且表面具有丰富PtZn-ZnOx界面的PtZn-ZnOx催化剂。像差校正高角环形暗场扫描透射电子显微镜(AC-HAADF-STEM)所摄图像和X射线衍射谱(XRD)证明了PtZn IMA的形成,其颗粒分布均匀,平均粒径为2.38 nm,测得的晶格间距(0.286 nm)符合金属间化合物Pt1Zn1沿[110]方向的晶格常数,说明PtZn IMAs中存在暴露的(110)面。
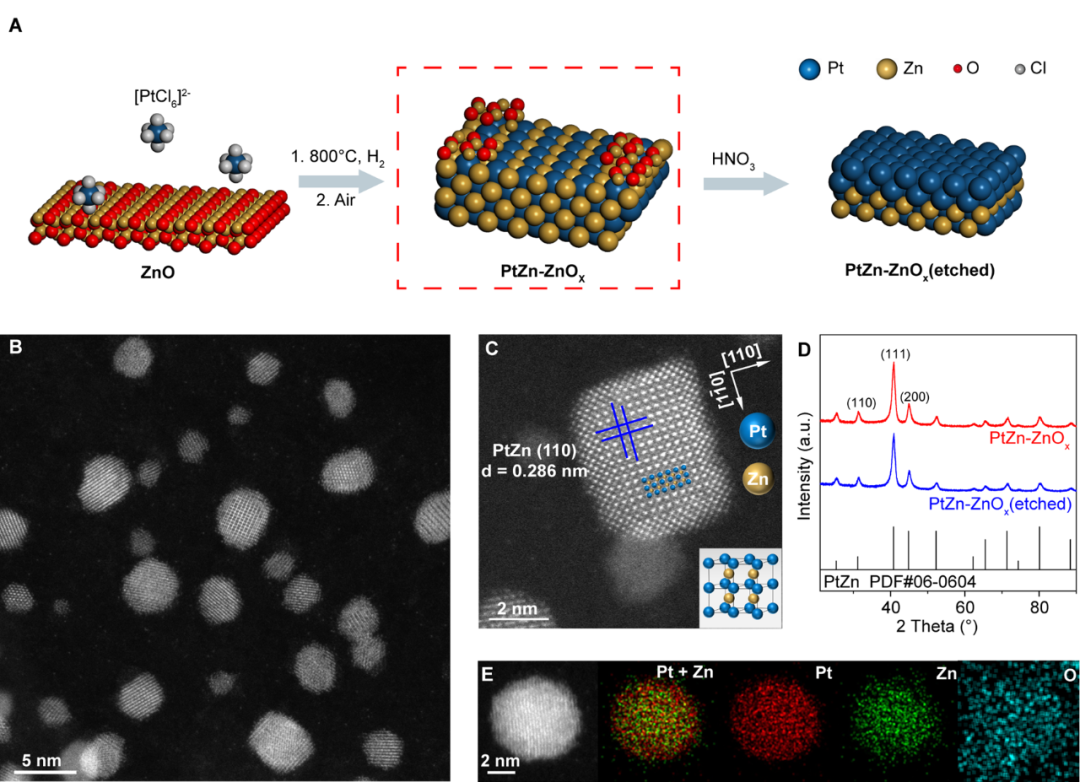
图1 PtZn-ZnOx催化剂的合成及结构表征。(A) PtZn-ZnOx和PtZn-ZnOx(etched)形成示意图。(B) PtZn-ZnOx的AC-HAADF-STEM图像。(C) PtZn-ZnOx纳米颗粒的AC-HAADF-STEM图像(插图为Pt1Zn1IMA的晶体结构图, Pt:蓝色;Zn:黄色)。(D) PtZn-ZnOx和PtZn-ZnOx(etched)的XRD图谱。(E) PtZn-ZnOx的AC-HAADF-STEM元素mapping。
作者利用X射线吸收精细结构(XAFS)和X射线光电子能谱(XPS)进一步研究了催化剂的化学状态和配位环境。研究表明:
(1)PtZn-ZnOx中Pt的价态高于0价: Pt L3-edge的白线峰强度高于Pt箔(图2A),Pt 4f7/2轨道的结合能相对Pt0发生正移(图2E);
(2)PtZn IMAs表面存在一层ZnOx亚单层:PtZn-ZnOx的Zn K-edge的吸收边能量显著高于Zn箔(图2B),在FT-EXAFS光谱中观察到1.6Å处存在一个Zn-O的峰(图2D),Zn L3M45M45的俄歇光谱中同时观察到Zn2+和Zn0的存在(图2F),此ZnOx是由PtZn IMAs表面的不饱和配位Zn原子在H2还原后暴露于空气中再次被氧化形成的;
(3)为了对比说明PtZn-ZnOx界面的作用,采用酸刻蚀法去除ZnOx制备得到PtZn-ZnOx(etched)催化剂,XAFS和XPS结果显示ZnOx已被去除。
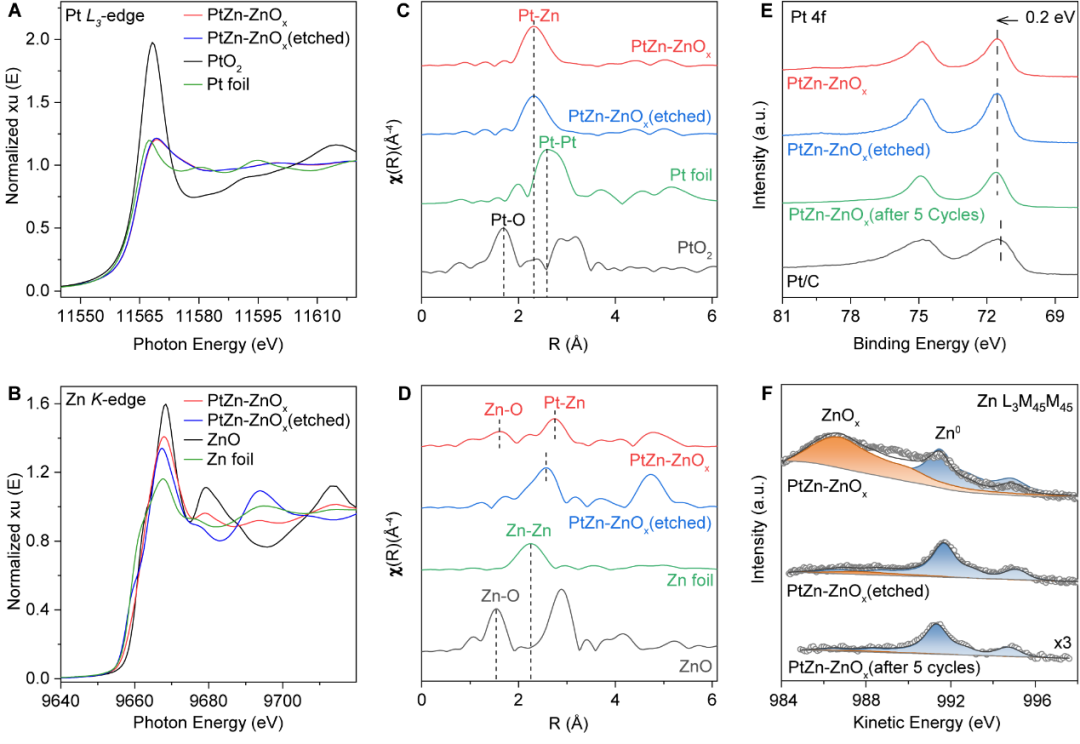
图2 PtZn-ZnOx和PtZn-ZnOx(etched)的化学价态和配位环境研究。PtZn-ZnOx和PtZn-ZnOx(etched)在Pt L3-edge(A)和Zn K-edge(B)处的XANES光谱。PtZn-ZnOx和PtZn-ZnOx(etched)在Pt L3-edge(C)和Zn K-edge (D)处的FT-EXAFS光谱。PtZn-ZnOx、PtZn-ZnOx(etched)和PtZn-ZnOx经过5次循环测试后的催化剂的XPS光谱,(E) Pt 4f轨道和(F) Zn L3M45M45轨道。
采用线性扫描伏安法(LSV)和计时电流法(CA)评估催化剂的催化剂性能,结果显示:
(1)PtZn-ZnOx催化剂可以在0.2 V(vs RHE)的低电压下驱动苯甲醇氧化反应的发生,比商业Pt/C低160 mV;在0.85 V(vs RHE)下的峰值电流密度为126 mA cm-2,电化学活性面积(ECSA)归一后的电流密度是商业Pt/C的2倍(图3A-B);
(2)PtZn-ZnOx催化剂可以在0.725 V(vs RHE)的低电压下实现高苯甲酸选择性(99.5%),比大多数报道的研究低0.6 V(图3D),选择性显著高于商业Pt/C(68.5%),并且在氧化过程中未检测到CO的生成;与其他已报道的BAOR催化剂相比,PtZn-ZnOx催化剂表现出更优异的性能,具有更低的起始电位和更高的峰值电流密度(图3H);
(3)酸刻蚀去除PtZn-ZnOx界面后,催化剂PtZn-ZnOx(etched)的BAOR电流密度下降到原来的80%,并且起始电位明显变高(图3A),这说明PtZn-ZnOx界面对促进PtZn-ZnOx催化剂上BAOR活性起着非常重要的作用;
(4)经过5次循环BAOR试验后(图3E),PtZn-ZnOx催化剂上的苯甲醇转化率(85%→72%)和Ph COOH选择性(99%→82%)略有下降。相比之下,PtZn-ZnOx(etched)催化剂在5个循环中保持了相对稳定的转化率(约75%)和选择性(约85%)(图3F)。这种现象是碱性溶液中PtZn-ZnOx催化剂表面ZnOx逐渐流失所导致的,进一步说明了PtZn-ZnOx界面对促进PtZn-ZnOx催化剂上BAOR活性起到的重要作用。
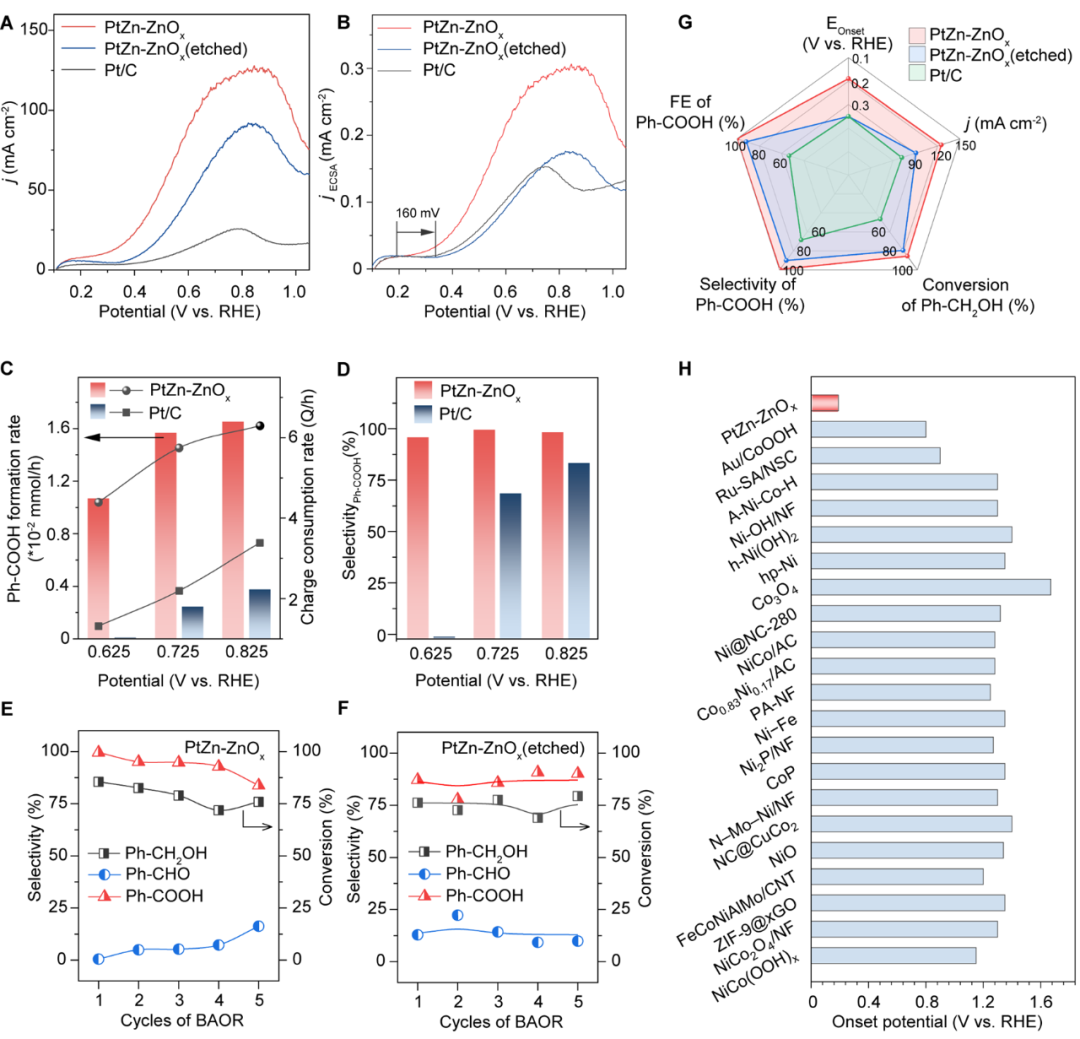
图3 催化性能及稳定性测试。(A)不同电催化剂在1.0 M KOH和0.1 M Ph-CH2OH条件下的LSV曲线。(B)不同电催化剂对BAOR的ECSA归一化LSV曲线。PtZn-ZnOx和Pt/C在0.625 V、0.725V和0.825V(vs. RHE)下在1.0 M KOH+5 mM Ph-CH2OH条件下的Ph-COOH的形成速率、电荷消耗速率(C)和Ph-COOH的选择性(D)。在0.725 V(vs. RHE)下,PtZn-ZnOx(E)和PtZn-ZnOx(etched) (F)上连续5次BAOR测试的选择性和转化率变化。(G)不同电催化剂的起始电位(EOnset)、峰值电流密度(j)、Ph-CH2OH的转化率、Ph-COOH的选择性和法拉第效率(FE)的比较。(H) PtZn-ZnOx催化剂与现有文献报道的催化剂BAOR起始电位的比较。
为了深入揭示PtZn-ZnOx催化剂上BAOR机理及C-H键活化机制,本研究进行了密度泛函理论(DFT)计算,构建了在PtZn(110)表面以及在PtZn(110)表面沉积一层ZnOx的ZnOx/PtZn表面来模拟PtZn-ZnOx催化剂的界面效应。计算结果显示:
(1)苯甲醇和苯甲醛在ZnOx/PtZn上的吸附能(Gads)均强于PtZn(110)(图4A),与开路电位(OCP)实验结果吻合良好(图4B);由于界面不饱和配位的ZnOx存在,ZnOx/PtZn对OH–的吸附更强;Bader电荷分析表明,界面配位不饱和Zn原子具有明显的正电荷,有利于氧化物的吸附;
(2)结合原位红外结果提出反应机理(图4C),反应开始于苯甲醇和OH*在催化剂表面的共吸附,然后逐步脱去-OH和C-H上的H并产生苯甲醛。随后的醛基(CHO)首先与OH*偶联,转化为中间体Ph-C(OH)HO*,然后与OH*发生C-H键断裂反应,生成苯甲酸(Ph COOH*);
(3)分别在ZnOx/PtZn和PtZn(110)表面上计算了BAOR反应路径的吉布斯自由能图(图4D),结果显示,ZnOx/PtZn和PtZn(110)的速控步(PDS)均为Ph CHO*与OH*偶联生成Ph C(OH)HO*的步骤,其ΔG值分别为0.49和0.59 eV,表明ZnOx/PtZn上CHO基团氧化的反应活性更高;
(4)上述结果证明了PtZn-ZnOx界面中的不饱和配位Zn原子促进了苯甲醇和苯甲醛的吸附和亲电OH*的生成,且PtZn-ZnOx界面降低了OH*与苯甲醛偶联的能垒,从而提高了催化活性和选择性。
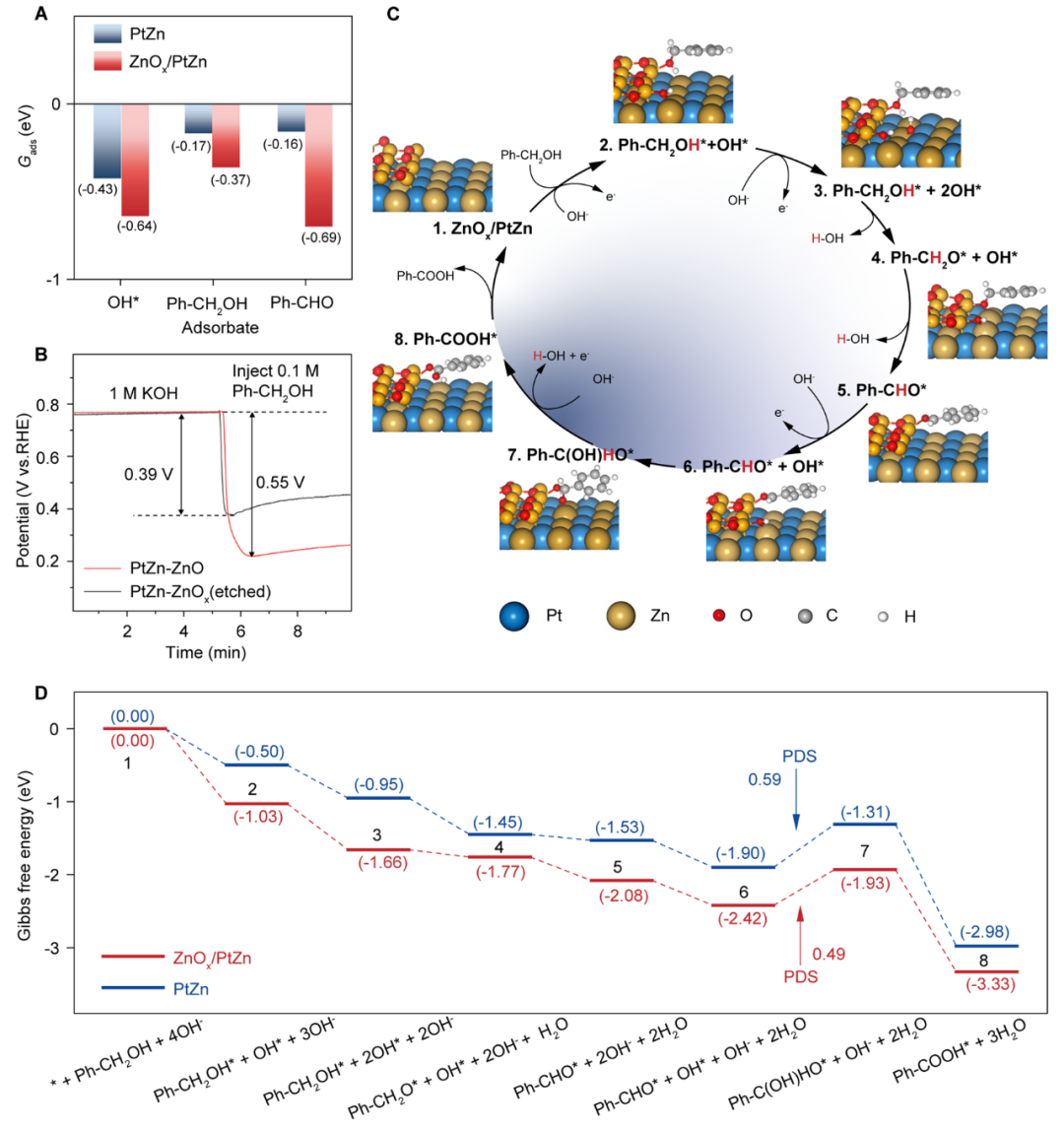
图4 BAOR在PtZn-ZnOx界面上的反应机理。(A) OH–、Ph-CH2OH和Ph-CHO在ZnOx/PtZn界面和PtZn表面的吸附能(Gads)。(B) 1 M KOH中快速注入0.1 M Ph-CH2OH时PtZn-ZnOx和PtZn的OCP曲线。(C)由DFT计算得出的ZnOx/PtZn界面BAOR反应过程示意图和相应的优化中间体几何形状。(D) Ph-CH2OH在ZnOx/PtZn和PtZn上氧化成Ph-COOH的吉布斯自由能图。括号里的数字是eV单位的吉布斯自由能。
本研究成功构建了具有丰富PtZn-ZnOx界面的金属间PtZn-ZnOx催化剂,成功实现在0.725 V (vs RHE)的BAOR低电压(比大多数报道的研究低0.6 V)下对苯甲酸的高选择性(99.5%)。实验结果和DFT计算突出了PtZn-ZnOx界面活性的增强,并阐明了内在的催化机理。具体来说,PtZn-ZnOx界面增强了反应中间体的吸附和活化,促进了亲电OH*物质的生成。降低了Ph-CHO与OH*偶联的能垒,保证了较高的催化活性。此外,PtZn-ZnOx在低电位下对多种醇类表现出良好的电催化氧化活性,证明了该催化剂的普适性。总而言之,这项工作为在低电位下高效电氧化有机小分子的催化剂合理设计,提供了一条可行的途径。
Renfeng Liu, Wenjing Tu, An Pei, Wei-Hsiang Huang, Yanyan Jia, Peng Wang, Daoru Liu, Qiqi Wu, Qizhen Qin, WeiWei Zhou, Linan Zhou, Keyou Yan, Yun Zhao, and Guangxu Chen. Low-Voltage Electrooxidation of Benzyl Alcohol to Benzoic Acid enhanced by PtZn-ZnOx Interface,J. Am. Chem. Soc. 2025.
https://doi.org/10.1021/jacs.4c17193
陈光需,华南理工大学环境与能源学院教授,博士生导师。2014年于厦门大学化学化工学院无机化学专业获得博士学位,师从郑南峰教授。2014年至2015年,在厦门大学能源材料化学协同创新中心从事博士后研究。2015年至2019年,在斯坦福大学崔屹教授课题组进行博士后研究。2018年入选海外高层次引进人才青年项目。主要研究兴趣是功能纳米材料的控制合成,纳米材料的表界面结构控制与表征以及功能纳米材料在多相催化和电催化性能的研究等。至今,在Science,Nat. Mater., Nat. Catal., Nat. Commun., ACS Nano,Small,ACS Catal., ACS AMI,EST,Nano Lett.等国际著名期刊上发表SCI论文60多篇。论文已被引用超过13000次(Google scholar),H因子44。
赵云,华南理工大学环境与能源学院副教授,博士生导师。2016年于厦门大学化学化工学院物理化学专业获得博士学位。2017至2019在德国莱布尼茨催化研究所进行博士后研究。2020.09至今担任华南理工大学环境与能源学院教学科研系列教师。研究方向为大气污染控制的理论研究,多相催化的功能纳米材料表界面调控,含碳小分子的活化及其催化反应机理的深度挖掘以及DFT理论模拟,机器学习在催化剂筛选方面的应用等。
裴安,现于华南理工大学环境与能源学院从事博士研究工作(导师陈光需教授),主要从事金属/载体界面电(热)催化性能及原位反应机理、电化学能源器件相关研究,近五年以第一作者/通讯作者在J. Am. Chem. Soc. (2篇), Nat. Commun. (2篇), Energy & Environ. Sci. (封面论文, ESI高被引), Adv. Funct. Mater.等期刊发表论文十余篇。曾获评硕士研究生国家奖学金,江西理工大学第十六届“十佳大学生”暨第十二届“泰豪之星”提名及江西理工大学优秀硕士毕业生,获评博士研究生国家奖学金,华南理工大学校长奖学金,厦门大学化学化工学院第七届博士研究生学术论坛二等奖等。
贾艳艳,华东理工大学讲师,2015年毕业于厦门大学,获理学博士学位,2014-2015在美国埃默里大学联合培养。2019年加入华东理工大学化学与分子工程学院,主要从事能源/环境催化剂构效关系研究。在J. Am. Chem. Soc.、Nano Letter, Chemical Engineering Journal等国际知名期刊发表学术论文20余篇。