

做实验的同学最想知道的。
1,我什么理论都不会,我可以做计算吗?
答:可以,不信你看完全文你就懂了。
2,计算需要什么准备?
答:这里只针对VASP计算,需要(机群or普通计算机or超算)
机群一般是指课题组自己买的,十几个GPU或CPU连在一起的设备,一般学校总会有几个课题组有机群。
普通计算机就是指你的台式电脑或笔记本电脑,不要求性能多好,只要可以上网打开浏览器就行,用来连接云计算平台。划重点:不需要自行安装Linux系统,Windows系统的电脑就能操作!
超算就是大科学装置,一般花点钱就能租到机时,他会给你个账户,账户里有一定时限的集群使用权,用完了就没了。
3,多少天可以学会计算?
答:一个小时or无限。
一个小时,是指你只要懂点高中数学,高中物理,一个小时你就能开始计算。
甚至利用一些现有的平台,十分钟你就可以开始计算。
无限是指计算这一领域博大精深,你永远可以往下挖,你永远可以有新知识学。
1 什么是计算?
计算其实和做实验没什么区别,一张图!


做实验,无非就是拿几个原料,丢到锅炉里面,烧结,然后得到目标化合物,测个xrd,得到结构信息。
做计算也是一样,只是这里的原料变成了具体的原子,锅炉变成了晶胞,烧结变成了结构优化,得到的最终结构同样可以算个xrd(软件一步生成!)。
具体解释一下:
原料:原子!
你丢入的可以是原子(也可以通过特定操作丢入离子),比方你想计算一个O2氧分子,你就往一个晶胞里面丢2个O原子。
锅炉:晶胞!
做材料的应该都知道晶胞的意思,这里的晶胞就是指重复单元!比如你构建一个很大的晶胞,里面只有一个O2氧气(如下图),那你要意识到,其实在他隔壁还是有氧气的。只是当我们晶胞构建的足够大,我们可以认为O2和相邻晶胞的O2的作用力太小,可以忽略。


烧结:结构优化!
结构优化就是计算的核心环节。
你丢两个氧原子的时候,位置是随意的,你可以让他们挨的近一点,也可以让他们挨的远一点,这是无所谓的,不管初始结构如何,结构优化后都会只有一个结果——两个原子构成最稳定的排布。
什么是结构优化?

图1 现在有两个氧原子,假设我们一开始放的距离偏大。两个氧原子之间吸引力大于排斥力,在力的作用下两个原子各靠近一步,变到图2。
图2 两个氧原子现在靠的太近了,之间吸引力小于排斥力,在力的作用下两个原子各远离一步,但是通过计算软件的算法,这一步移动的距离是小于上一步的,又回到图1。
图3 不断重复1和2,原子的距离不断变换,直到吸引力和排斥力近乎相等。这时我们会设置一个界限,比如当(|吸引力-排斥力|
注:实际计算的界限肯定不是1牛顿,而是一个远小于这个的值!
结构优化后的数据分析
现在结构优化完,我们也得到我们的目标结构了,(比如任意距离两个氧原子结构优化后,我们会发现两个氧原子的距离刚好就是氧气分子的键长,既他们结合构成了氧分子。)那我们现在可以拿这个目标结构来干嘛?
1.首先,我们可以分析结构的基本信息,比如我们可以得到O2的键长,电荷分别等。如果是晶体材料,我们还能得到其他结构信息,如晶格参数。
2.我们还可以电子分布信息,可以进一步由这提取出化合价,电子分布,成键方式,能带,带隙。
看到这里,恭喜你,你已经掌握计算的基本流程。目前你还有两个技能树可以拓展:
1.原子及电子相互作用力计算方法。
2.具体计算操作细节。
力是怎么算的,这就涉及一个复杂的问题了,涉及到原子的势场,原子的电荷分布,需要用到第一性原理理论的东西,这也是第一性原理计算名字的由来,想了解到可以去看一下凝聚态物理和密度泛函的书籍,不想看的就把他简单地想像成“高级”的牛顿力学计算。
如果不想了解,也没关系,你就把他当成个工具用就行,就像你烧炉子的时候,会去纠结炉子内部电路原理吗?
2 计算具体操作
首先,我们拿什么软件?
目前主流的晶体结构的计算,用的是VASP。
VASP安装在哪?
如之前所述,你有(机群or超算or云计算平台)三个选择。
机群是最麻烦的,得在Linux系统上完成软件的安装编译,运行,读取结果。对于不了解Linux系统的人非常不友好。
超算是比较简单的,他们应该都已经帮你装好了,你只管用就行。
云计算平台是最简单的,而且是Windows系统界面操作就行,只需要链接互联网打开浏览器就行。用浏览器打开云计算的网站,计算所需要的软件平台都帮你弄好了,直接开始算就行。
有人可能不太懂超算和云计算平台的区别。首先,两者底层原理是一样的,都是你用你的电脑远程连接集群(超算中心)的核心电脑。两者区别在于,超算你连接后,操作界面是Linux界面,云计算平台连接后,操作界面是Windows的浏览器界面。
另外,可以把VASP计算结果可视化的软件是VESTA。
【免费课程】快速精通VESTA建模:异质结、纳米线、量子点、表界面、体相晶体模型等!
有了软件,下一步就是运行!
运行前需要准备工作,就是四个文本文件。
这四个文件是告诉软件,你要算什么?你要怎么算?你想得到什么结果?
POSCAR,POTCAR,KPOINTS,INCAR
第一:POSCAR
POSCAR(position + car):结构车厢。
里面放着就是初始结构的位置。
实例:
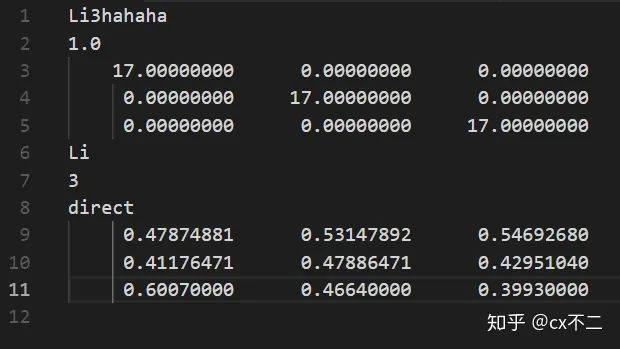
第一行就是名字,无所谓,可以随便取。
第二行是1.0,晶胞的缩放系数,别管那么多,反正1.0就对了。
第三到第五行,晶胞参数,不懂的自己去翻材基课本。
第六行:晶胞内元素种类,这里是Li,示意锂元素。
第七行:原子个数,3,示意三个锂原子。
第八行:不用管,direct就对了。
第九行到第十一行:三个原子的具体坐标。
展示以下具体的结构示意图,可用vesta查看。



第二:POTCAR
POTCAR是用来描述原子的物理性质的,比如晶胞内有O原子和Li原子两种原子,他们的性质肯定是不一样的,你总不能只给出一个是O一个是Li后就什么也不管了吧。
还得给出原子的具体信息,如质量,电荷量,电荷分布什么的,这些信息很复杂。
这个文件,也被称为赝(yan)势,赝就假的的意思,势指势场,赝势就是指假的势场,在这里的意思是模拟的势场,它和真实势场存在一定误差。
第三:KPOINTS
K点文件,具体涉及布里渊区什么的,入门阶段,你只需要知道你取的点越密,计算精度越高,耗时越长。
实例:
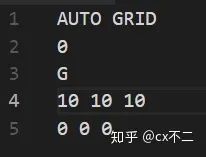
入门阶段,你唯一需要改动的就是第四行,就是K点,具体取多少,没有硬性规定,打个比方,你可以取10 10 10,也可以取 5 5 5。但是 10 10 10 肯定比 5 5 5 算的精度要高,但是速度也慢!
第四:INCAR
INCAR里放的是计算参数,比如精度,计算步数。
里面的参数很多,对于初学者,你只需要知道我给出的下面8个,然后再慢慢拓展。
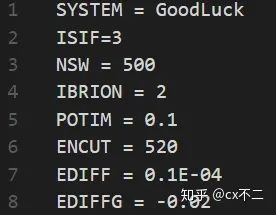
第一行 SYSTEM 就是计算体系的名词,随便取。
第二行 ISIF 就是设置优化的参数,一般取2或者取3。2代表结构优化过程,晶格参数不会变;3代表晶格参数会变。两者有什么区别呢?如果你是在优化某一晶体,你给的初始晶格参数不是最稳定的,你就必须得用3,让他自己找到最稳定的经过参数。但如果你是在计算某一气体分子,那你就可以取2。
第三行 NSW 最大优化步数,在结构优化中,每移动一次原子我们称为一步。我这边设置NSW=500,意味着如果原子移动了500步,结构还没有达到我给的稳定条件,那么也停止计算。一般500步还不收敛,那结构可能就有问题了,也没必要继续计算。
第四行 IBRION 就是原子移动的算法,一般取2就行,如果是分子动力学,则需要取别的值。
第五行 POTIM 原子移动步长。尽管我们不知道具体的算法,但是我们应该意识到,移动的dx和受力F应该是有个递增关系的:dx=k*F,k是个系数。如果k越小,每步原子移动的越慢。这个POTIM就是和K有关,POTIM越大K越大。
第六行 ENCUT 截断能,具体意思自己百度,对于初学者,你只需要知道截断能越大,计算越准确,耗时越长。一般520满足绝大多数体系的计算。
第七行 EDIFF 能量收敛标准,当相邻两步的能量差小于设定值,我们就认为能量稳定,这里取得是0.1E-04 eV。
第八行 EDIFFG 力的收敛标准,当体系中任意两个原子的相互作用力的最大值小于设定值,我们就认为结构稳定,这里取得是 0.02 eV/A。
除了这些参数,还有ISPIN,ISTART,ISYM,ISMEAR等参数需要初学者知道,不过你可以先用之前的八个参数,完成简单的计算。
VASP是备受专业技术人员认可的DFT计算软件,每年发表论文超过1万篇,其计算结果不仅可以验证实验,更能够预测实验结果,指导实验路线。
“实验+计算”的模式已成为顶刊标配!为了让广大科研人员能够尽快上手VASP,掌握顶刊核心技能,华算科技朱老师原创设计了VASP系列课程!
七大VASP专题课程,带你从入门到进阶,玩转DFT计算!晶体、二维材料、催化、电池、钙钛矿、单原子、吸附、半导体、缺陷计算等培训汇总!
两个课程同时报名可优惠1600元,多报享更多优惠!
报名方式:添加下方微信好友报名,手机:13128723011。
👇👇扫描二维码,立即报名👇👇
