



尿素电氧化是一种经济有效的替代水氧化的节能制氢方法。但其实际应用受到尿素反应物昂贵和反应动力学缓慢的限制。
阿德莱德大学乔世璋院士、郑尧教授等人提出了一种高效的尿液电解制氢系统,使用成本免费的尿液作为原料。该系统利用了在Pt催化剂上发现的Cl介导的尿素氧化机制,其中吸附的Cl直接与尿素偶联形成N-氯脲中间体,然后通过分子间N-N偶联转化为N2。这种快速的介质氧化过程显著提高了尿液电解的活性和稳定性,同时避免了Cl引起的腐蚀,可以在低电压下运行200小时以上。因此,在实际的制氢电解槽中,在300 mA cm-2下电解尿液(4.05 kWh Nm-3)的电耗显著降低,优于传统的尿素(5.62 kWh Nm-3)和水(4.70-5.00 kWh Nm-3)电解。
相关工作以《Urine electrooxidation for energy–saving hydrogen generation》为题在《Nature Communications》上发表论文。团队成员王鹏棠(博士毕业于苏州大学)为论文第一作者。
值得注意的是,从Peer Review文件上可以看到,该研究论文在投稿阶段曾受到了两位审稿人的质疑。审稿人1认为:虽然研究工作是充分的,效果是令人满意的,但除了作者提出的特殊机制外,所设计的系统并不显得“新颖”。
例如,在纯实验室条件下对中性、酸性、碱性条件下尿素氧化的对比研究具有一定的意义,但对于实际工业废水来说,其来源复杂,各种条件都要有相应的对策。具体来说,对于尿液废水,以中性或微碱性为宜,将尿液重新转化为强酸性条件进行处理,这在工程实践中并不是理想的选择。
工业尿素废水中尿素浓度较高。但如何保证尿废水中尿素含量稳定(~0.33M)?毕竟,厕所里的尿液流向污水处理厂,显然是被稀释了很多倍。即使是收集到的纯净的新鲜尿液,在储存后也很容易水解成氨。此外,应测量每种情况下的实际尿液处理情况。
另一位审稿人也提出了一些担忧:令人遗憾的是,这项研究只使用了合成尿液,而没有实际的原始尿液的结果。作者分别在酸性和碱性条件下使用贵金属Pt/C电极和廉价的NF电极进行尿液电解。这种比较似乎不公平。

针对上述意见,作者也是积极给出了回复:不同于以往的Cl-介导电解研究依赖于添加尿素和含Cl盐作为反应物,本文的系统直接利用模拟尿液甚至原尿作为电解的原料,系统地评估尿液的各种参数对Cl-介导电解的影响,包括杂质离子、pH、尿素浓度和储存条件。这为尿液电氧化及利用系统的设计提供了新的思路。对于选择强酸性条件进行尿液处理和电解的原因如下:
(1)酸性条件是通过Pt催化剂驱动Cl介导的UOR的必要条件,克服了在碱性介质中Ni催化UOR时观察到的Cl诱导活性和稳定性衰减的问题;
(2)与碱性或中性尿液废水相比,酸性条件在动力学上更有利于阴极HER,也有效地降低了氯介导的阳极反应的过电位,这有助于通过尿液电解更有效地节能制氢;
(3)在尿液储存过程中,酸性环境防止尿素水解,保持尿素含量,并确保有足够的反应物用于制氢。此外,作者也承认审稿人的担忧,即强酸性条件可能不适合实际工程应用。为了解决这个问题,作者在弱酸条件下测试了尿液电解。

此外,审稿人除了对文章提出体系保留疑问外,还对反应机制、作用机理也提出了进一步的担忧。例如,由于存在游离氯,与尿素-N的反应可能涉及氯胺中间体的形成。

最后,作者总共用了42页PDF仔细回复了审稿人提出的质疑(正文对于反应机理作出了详细的解释),最终成功发表论文。

图1 电解尿液制氢的现状
通过技术经济分析(TEA,图1a),考虑到尿素的市场价格和所需电压,使用纯尿素进行UOR是不切实际的,在节省成本的制氢方面很难与电解水竞争。进行UOR以节省成本的氢气生产的唯一可行途径需要无成本且丰富的尿液作为原料。然而,除了足够量的尿素外,人体尿液中还含有镁、钙和氯离子。这些额外的成分通过形成沉淀或中毒催化剂对碱性UOR产生负面影响。特别是,在工业规模的电流操作下,尿液中相对大量的Cl−离子会引发氯析出反应(ClER),导致电极和电解槽的不可逆腐蚀。此外,在Ni催化的UOR下,尿素主要转化为有害的亚硝酸盐(NO2–)。由于NO2–比水更容易还原,在电场作用下,NO2–会向阴极迁移,与析氢反应竞争,导致产氢电流效率下降(图1b)。
受益于Cl–/Cl2的低热力学平衡势,酸性尿电解提供了通过触发Cl2辅助尿素氧化(6Cl–→3Cl2+6e–;3Cl2+CO(NH2)2+H2O→CO2+N2+6HCl),理论上有助于降低H2生成的总体潜力(图1c)。然而,由于传质的限制,这种在Cl2存在下的间接尿素氧化表现出缓慢的动力学和潜在的腐蚀,导致低电流密度和令人失望的尿电解稳定性(-2,图1d)。这种较差的性能不能满足工业规模的要求,严重增加了制氢成本(图1e),迫切需要通过挖掘替代中间体的反应机理和加速动力学来改进。
本文提出了一个动力学有利的Cl介导的UOR(表示为Cl-UOR)途径,用于具有成本效益的制氢。与涉及Cl2生成和参与的缓慢间接氧化机制不同,本文证明了通过吸附的Cl(Clads)与吸附的尿素直接偶联形成N-氯脲可以在Pt表面加速Cl-UOR,然后通过分子间N-N偶联在*OH的帮助下转化为N2(图1f)。这种尿电解中Cl介导的氧化途径/机制避免了Cl2的产生,有助于提高该过程的性能。

图2 碱性和酸性条件下模拟电解尿液的比较
在双电极H型电解池中,首先评估了纯尿素和模拟尿液作为电解添加剂在碱性和酸性电解过程中的差异。如图2a所示,在尿素溶液中加入相应浓度的金属盐制备模拟尿液。考虑到以往尿素电氧化研究中常用的电极是泡沫镍(NF)和铂/碳(Pt/C),因此选择它们作为碱性和酸性电解的电极。LSV测试表明,在含1 M KOH的纯尿素中,NF在电位1.3 V时电流密度增加,具有良好的尿素电氧化活性(图2b)。然而,一旦用模拟尿液代替纯尿素,NF活性在高电位下明显下降,并在1小时内出现明显的电流衰减。在高活性的Ni(OH)2/NF电极上也观察到类似的行为,表明模拟尿液中Cl–不可避免地在高电位下腐蚀Ni基催化剂,从而导致UOR期间的活性损失。
相反,铂/碳催化纯尿素或模拟尿液在酸中电氧化显示相反的结果。在0.5 M H2SO4中,当加入纯尿素时,Pt/C电流没有明显增加,而在用模拟尿液代替尿素后,电流有显著提高。与NF相比,在0.5 M H2SO4条件下,以模拟尿液为添加剂的UOR在2 V下的Pt/C活性可达到118.9 mA cm-2,是纯尿素的13倍,且在1 h内保持良好(图2c)。所有这些结果都表明了镍基催化剂在尿电解制氢方面的性能局限性,与Pt/C催化制氢过程中所观察到的性能增强形成了对比。
为了研究模拟尿液中UOR增强的潜在因素,分别评估了模拟尿液中存在的各种离子对Pt/C UOR性能的影响。如图2d所示,在酸性条件下,尿中除Cl–外,其他离子的存在对UOR中Pt/C的活性没有显著影响,证实尿中Cl–对UOR的促进起着至关重要的作用。然后探讨了酸性条件(pH)对Pt/C的UOR改善的影响,发现微酸性电解质(pH值为1或2)有望用于实际的尿液电解,平衡性能与节能制氢的工程可行性。此外,对照酶解实验表明,与未酸化尿液相比,酸化尿液可以阻止酶解尿素,在储存期间保持稳定的尿素浓度(图2e)。因此,酸化储存尿液的UOR活性仍然很高,与新鲜尿液相当,而未酸化储存尿液的UOR活性可以忽略不计(图2f)。所有这些结果都证明了Cl–和酸性条件在促进Pt催化尿素氧化制氢中的关键作用。

图3 酸性条件下Pt催化Cl-UOR的产物分析
本文采用原位差分电化学质谱法(DEMS)研究了Pt催化尿液在酸中电解的可能途径。在含KCl的电解液(0.5 M H2SO4)中,Cl2的检测信号在-1.35 V 下逐渐增加,表明触发了ClER。然而,当向该电解质中加入尿素时,先前观察到的Cl2消失,并且可以看到1.5 V下CO2和N2增加的信号(图3a)。更重要的是,ClER平衡电位(1.36 V)范围内的可控CV扫描显示,一旦尿素被额外添加到电解质中,除了Cl2还原峰外,定制扫描范围内的Clads解吸峰也会消失(图3b)。这些结果表明Clads在Pt上作为Cl2以外的活性介质。
在电解1h后,NF催化的碱性UOR和Pt催化的酸性Cl-UOR生成的每个产物的法拉第效率(FEs)的潜在依赖性总结在图3c、d中。正如预期的那样,在低电位下,N-氯脲是主要产物,随着电位的增加,N-氯脲逐渐被N2取代。与NF上的UOR相比,Pt/C上Cl-UOR的N2的FE在1.9 V时达到最大值73.1%(图3c、d),表明Pt上酸性Cl-UOR的优势,这有助于尿素转化为N2。
进一步研究了1.8 V下Cl-UOR的活度、反应物和生成物随时间的变化(图3e)。我们观察到,在电解过程中,电流密度逐渐降低,这可以归因于尿素和Cl–毒化的消耗。然而,与上述电解1小时的结果不同,随着电解时间的延长,溶液中游离Cl的浓度增加。当尿素浓度降至-0.074 M时,游离Cl浓度显著升高。这一结果凸显了UOR和ClER之间的竞争,随着尿素的消耗,ClER将变得更占优势。长期电解过程中,随着尿素用量的增加,N2的FE增加,而氯脲的FE降低,表明氯脲在Cl-UOR过程中起到二次反应物的作用。NH4+及其电氧化衍生物NO2–随着电解时间的延长而逐渐增加。这一发现表明氯胺可能在反应过程中作为中间体短暂形成,但不会作为最终产物持续存在,因为它不稳定,在酸条件下容易转化为N2。

图4 Cl-UOR催化剂性能评价及动力学研究
为了深入了解Pt上的Cl-UOR,选择了RuO2催化剂进行比较,因为它在ClER中的活性很高与观察到的对Pt/C的活性促进不同,与ClER相比,RuO2对Cl-UOR的活性要低得多(图4a),证明了Pt在促进Cl-UOR过程中的独特性。通过将Cl的浓度与活性联系起来进行动力学研究。如图4b所示,CCl-对Cl-UOR活性的依赖性在RuO2上很明显,线性斜率为1.25,而在Pt上为零,表明与RuO2相比,Cl-UOR在Pt上的动力学发生了改变。然而,Pt/C的情况更为复杂,呈现出两个阶段(图4c)。当CCl-低于0.5 M时,Cl-UOR和ClER的活性关系均为线性关系,斜率分别为0.65和0.78。然而,当CCl-增加超过0.5 M时,ClER线性活性保持斜率,而Cl-UOR趋于零。
更重要的是,与ClER相比,Cl-UOR的活性受Cl–传质的影响较小,但它明显随尿素浓度的变化而变化,在Pt/C上呈火山趋势,进一步证明了尿素对Cl-UOR动力学的重要作用。因此,推测尿素同时吸附在Pt表面与Clads结合,从而加速了Cl-UOR的动力学。为了验证这一推测,探索了Pt/C上氢的欠电位沉积(HUPD)。与0.5 M H2SO4中Pt/C的CV相比,尿素或Cl–的存在导致HUPD电荷减少,如图4d所示。此外,在尿素或Cl–存在的溶液中,观察到OH–吸附电位的增加这些结果为尿素和氯在铂表面的吸附提供了依据。此外,在Cl 2p 的XPS中发现了另外一对属于有机Cl的峰,与仅在KCl电解质中观察到的峰相比,在KCl/尿素混合电解质中反应后,这些峰随着施加电位的增加而增加(图4e)。由于XPS测试选择的反应电位(1.3 V)没有触发ClER(图4e),因此排除了Cl2与尿素反应产生的有机Cl物种,从而证实了吸附尿素与Clads的偶联。
总之,上述结果说明了在Pt/C上引发的Cl-UOR经历了一个快速氧化过程,尿素和Cl同时吸附在Pt表面进行直接耦合(图4g),这不同于不同于Cl2在RuO2上参与的缓慢间接氧化(图4f)。
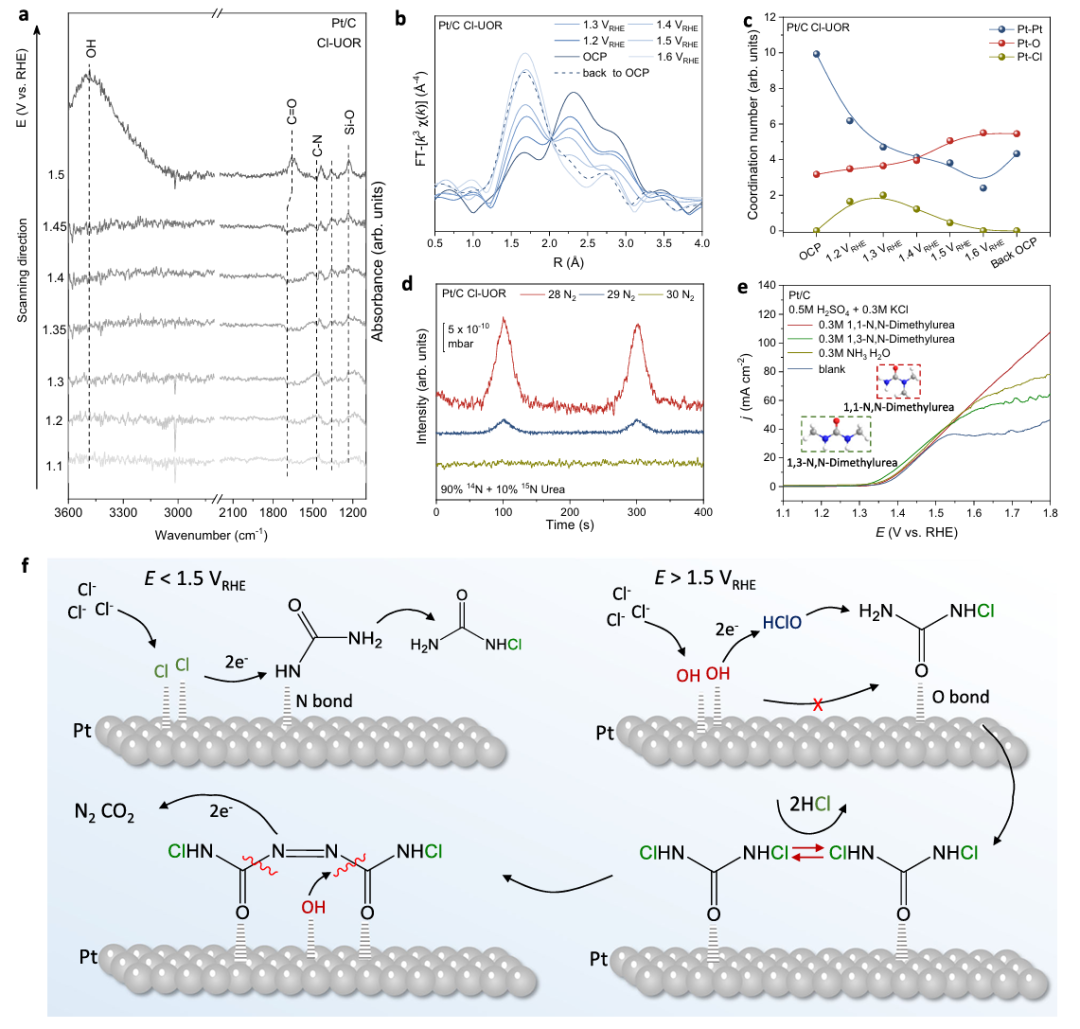
图5 通过Cl-UOR实现尿素转化为N2的机理研究
为了进一步阐明促进Pt/C上尿素-to-N2转化的潜在原因,通过原位ATR-IRAS评估了Cl-UOR过程中中间体的电位依赖性吸附。当电位增加到1.5 V时,C=O振动峰反转并移至~1656 cm-1,在~3490 cm-1处出现新的OH–振动峰,表明N-氯脲和*OH开始通过O端构型吸附在Pt表面。这一发现与1.5 V条件下发现的N2生成有关,提示由于电位诱导的局部环境变化改变了Cl-UOR的反应途径,这是尿素转化为N2的关键。
本文采用原位XAS研究了Pt在Cl-UOR过程中的价态和配位环境变化。对Pt/C的Pt的L3边缘EXAFS谱分析发现,在Cl-UOR过程中,电位从开路电位(OCP)上升到1.6 V(图5b),从Pt-Pt到Pt-Cl和Pt-O的初级散射峰发生了明显的转变,表明Pt配位结构在整个Cl-UOR过程中发生了动态变化。Pt-Pt键和Pt-O键的配位数(CN)分别随施加电位的增大和减小呈一致趋势,而Pt-Cl键的配位数则呈火山趋势(图5c)。特别是,在Cl-UOR期间,随着CNPt-Cl在1.5 V以上降至零,CNPt-O的增加是可见的,即使在电位返回到OCP时也几乎保持不变(图5c)。CNPt-O和CNPt-Cl的这种明显变化与FTIR光谱上1.5 V处出现的OH–吸附峰密切对应,表明由于Pt被吸附的OH–氧化而导致包层覆盖率下降。
考虑到这一发现,原来的Clads与吸附尿素结合的步骤被抑制,取而代之的是被吸附的*OH与溶液中的Cl–相互作用生成HClO。如前所述,盐水电解质的Pourbaix图可以证明这种电位依赖性的HClO生成。因此,所有这些结果表明,HClO成为1.5 V以上的Cl-UOR的主要介质,在尿素制氮中起重要作用。以上结果表明,在1.5 V以上,HClO成为Cl-UOR的主要介质,在尿素制N2过程中起重要作用。如图5d所示,以14N尿素(90%)和15N尿素(10%)混合物为反应物进行Cl-UOR试验时,在DEMS上仅检测到14N15N和14N14N信号,而未出现明显的15N15N信号,说明分子间N-N耦合是Cl-UOR过程中N2生成的主要途径。
此外,尿素类似物的可控电氧化表明,1,1-N,N-二甲基脲在1.5 VRHE以上Pt/C上的活性高于1,3-N,N-二甲基脲(图5e),表明伯胺部分对高电位活性促进至关重要,这是分子间N-N偶联的必要条件。此外,与尿素相比,氨在高电位下的活性要低得多,这排除了氨在Cl-UOR过程中由于尿素水解而发生分子间N-N偶联的可能性(图5e),并证明了在分子间N-N偶联步骤之后发生了C-N裂解以释放N2和CO2。
基于以上分析,提出了Pt/C上升压Cl-UOR的整个机理(图5f)。在低电位下,尿素通过N端构型吸附在Pt表面,并伴有包层,导致N-氯脲通过直接偶联生成的快速动力学。随着电位的增加,由于Pt的表面氧化,*OH开始吸附在1.5 V以上的表面。这种改变的吸附行为导致HClO的形成作为氧化还原介质,促进N-氯脲氧化成N,N’-二氯脲。随着N,N’-二氯脲在Pt表面覆盖面积的增加,分子间N-N偶联通过破坏C-N键,在*OH的帮助下自发触发生成N2和CO2,从而实现尿素到N2的转化。

图6 Pt催化电解尿液与电解水的比较
为了评估Pt催化的Cl-UOR作为OER制氢替代品的可行性,组装了一个流动型电解槽,使用负载Pt/C的碳布(CC)作为阳极和阴极,由PPS隔膜分开。与此相对应,为了进行比较,采用了与商业催化剂装配方式相同的两种类型的水电解槽(图6b)。如图6c所示,随着应用电位的增加,与酸性和碱性电解相比,Pt催化的模拟尿液电解显示出产氢电流密度的显著提高。此外,观察到更高的稳定性,确保在10 mA cm-2的电流密度下稳定运行210小时,用于制氢(平均FE为98.5%),大大超过相同条件下的酸性电解(图6d)。

图7 铂催化电解尿液与其它尿素(尿液)电解体系的比较
为了满足大规模制氢的工业要求,电解温度提高到60℃。结果表明,Pt催化的模拟尿电解在60℃下得到了进一步的改进,在2 V下电流密度为341.4 mA cm-2,分别是Ni(OH)2催化尿素和碱模拟尿电解的1.15和1.54倍。该值满足了工业规模电解的要求,这在以往的尿液电解研究中没有得到评估和实现,并且与本研究中电解水、氧化钌催化的尿液电解相比(图7a)也更高,突出了系统的节能优势及其制氢的实际潜力。
值得注意的是,在Pt催化的模拟尿液电解过程中,通过额外引入生理盐水将Cl–的浓度调至0.5 M,可以实现更高的活性,例如,~531.7 mA cm-2,这进一步证明了其在工业操作中的适用性(图7a)。此外,Pt表现出更强的稳定性,在模拟尿液电解超过20小时的操作和300 mA cm-2的10个循环中,Pt的平均电解电压保持在2.23 V(图7b),而Ni(OH)2和RuO2在相应的电解质中表现出严重的活性衰减,归因于Cl2腐蚀。由于Pt催化的模拟尿液电解具有较高的N2的FE,大大缓解了Ni基碱性电解制氢过程中NO2–引起的电流效率下降。因此,Pt催化模拟尿液电解的H2的FE得到了显著提高,达到~94%,超过了碱性条件下Ni(OH)2的~85%。
除了评估模拟尿液的电解外,电解系统还进一步使用直接从作者处批量收集的原始尿液作为原料进行操作。为了减少电解过程中有机物质对电极的污染,原始尿液样品经过酸稳定处理,并使用PTFE膜(0.22 μm)过滤器进行预处理,以去除有机杂质和大分子。如图7d所示,在不同批次的原始尿液样本中观察到Cl-UOR活性的变化,这归因于基于饮食摄入水平的有效成分(Cl–和尿素)浓度的差异(图7c)。当作者消耗更少的水和更多的盐来控制代谢时,生成的尿液(样本4)的Cl-UOR活性显著增加,达到与模拟尿液相当的水平(图7d)。
在原料尿液中加入低成本的粗盐(表示为样品1,0.2 M Cl–)也可以获得类似的结果,这表明尿液中Cl–对Cl-UOR增强的重要作用。此外,在60℃的测试温度下,原始尿液的Cl-UOR活性与模拟尿液相当(图7e)。这些结果进一步验证了系统在实际应用中的可行性。根据所得氢气FE和电池电压计算了各电解制氢系统的电费和能效(图7f)。令人印象深刻的是,Pt催化的模拟尿液电解确保了在300 mA cm-2操作下显着减少电力和提高H2生产的EE(80.1%)。产氢的电费达到4.42 kWh Nm-3,通过提高Cl–浓度可优化至4.05 kWh Nm-3。
Urine electrooxidation for energy–saving hydrogen generation,Nature Communications,2025. https://www.nature.com/articles/s41467-025-57798-3