MS锂电池电解质计算课程新增以下内容,学员可直接享有!包括:
1. 0.5小时视频:讲解顶刊文献常用的自由溶剂比例计算方法
2. 新脚本:自由溶剂分子比例、锂离子配位溶剂分子比例计算
作为横跨多个尺度的集成式材料计算模拟平台,Materials Studio在电池材料研究方面具有天然的优势。电池材料包括正负电极与电解液,其分子水平的设计同时需要量子力学与分子力学两大类方法,而MS的众多计算引擎模块恰能满足此要求。
除此之外,MS强大的建模功能和可视化界面对初学者极其友好,使初学者即便对于复杂的电化学计算也能够手到擒来。
本次锂电池电解质计算课程由华算科技和Materials Studio官方代理深圳浦华联合举办,基于Forcite,DMol3, Amorphous Cell,CASTEP模块设计,既包含液态、固态电解质的设计,也涵盖溶剂分子的电子性质研究。
此外,杨老师针对电解液开发了8个脚本,使分子动力学分析变得“一键即成”,使不会编程的小白也能上手复杂的电解液计算。
29小时高强度课程,理论讲解/实操建模/线上课程/无限回放/永久答疑!
课程主要知识点:分子动力学+AIMD、电解液建模、固态电解质、RDF+配位数、SSIPs+CIPs+AGGs、自由溶剂占比、MSD+离子电导率、离子迁移孔道+势垒、溶剂化效应+溶剂化能、HOMO/LUMO+静电势。
添加下方微信好友报名,或者联系手机133-1680-8231 。
注:此课程为MS电池系列课程之电解质计算培训,搭配锂电钠电正负极材料计算培训食用更佳!(点击链接跳转)与锂电钠电课程结合便宜多少钱?详情请咨询华算科技-嘉怡。
更多理论计算课程推荐:
Python/机器学习全系列课程:Python数据分析、机器学习与材料/化学、神经网络、机器学习与电催化等!
Materials Studio全系列课程:MS+LAMMPS建模/催化/电池/半导体计算培训…零基础到进阶!
Gaussian零基础入门培训:量子化学与建模、电荷、单点能、静电势、过渡态、IRC、势能面、光谱计算、溶剂化等!
VASP计算全系列课程:14大专题,金属、晶体、二维材料、催化、电池、钙钛矿、单原子、吸附、磁性、半导体缺陷计算等!
杨老师:华算科技全职技术资深专家,拥有14年以上Materials Studio软件使用经验,全网粉丝10万+。
曾就职于德国马克思普朗克研究所,日本WPI研究所,并曾在芬兰阿尔托大学进行长期访问,作为PI主持欧盟与日本科研项目各2项,日本高校科研项目2项。
主要从事固态相变的第一性原理研究、电化学固液界面的AIMD研究。
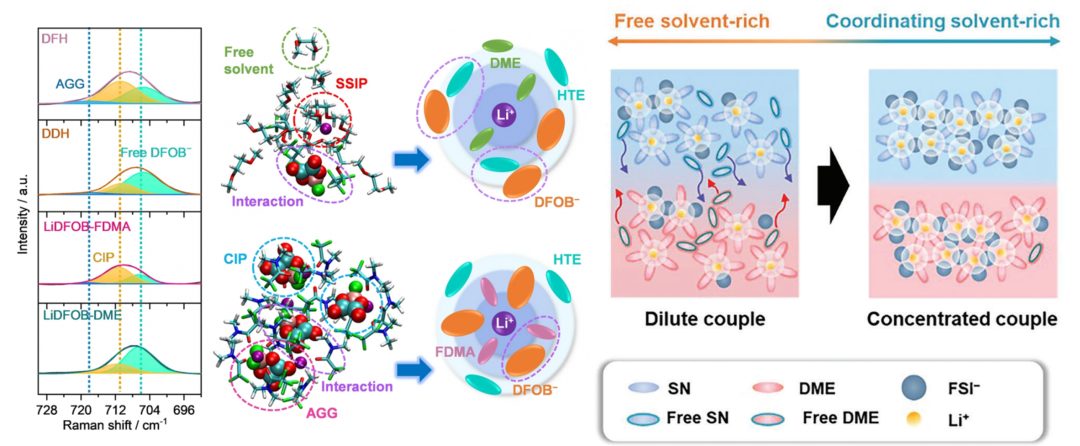
分子动力学、AIMD、电解液建模、固态电解质、RDF、配位数、SSIPs+CIPs+AGGs、MSD、扩散系数、离子电导率、离子迁移孔道、势垒、瓶颈、PF6–/TFSI–、溶剂化能、溶剂化效应、HOMO/LUMO、静电势、电荷布居、福井函数。
✅体系全面化(多种液态、固态电解质)+手段系统化(分子动力学+AIMD+DFT计算)
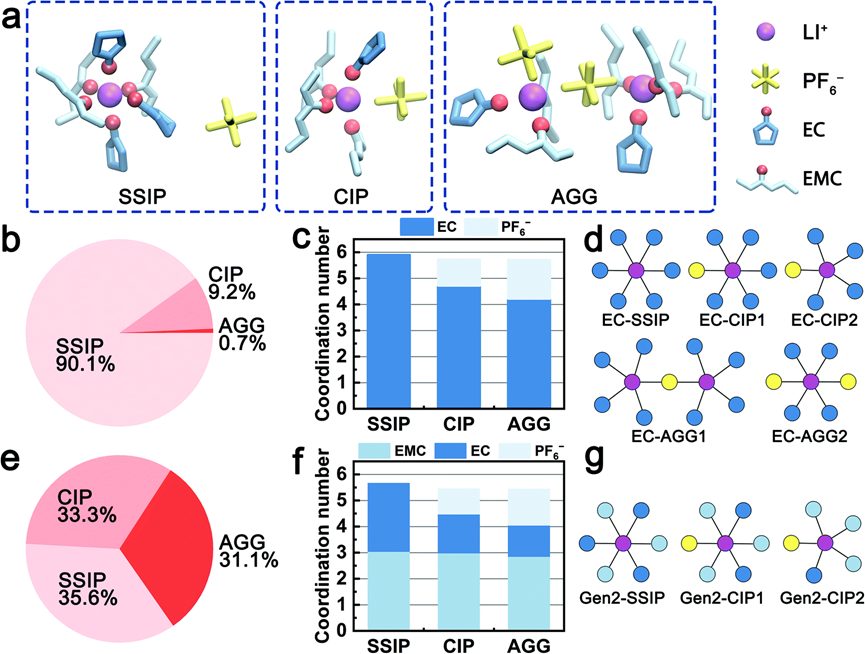
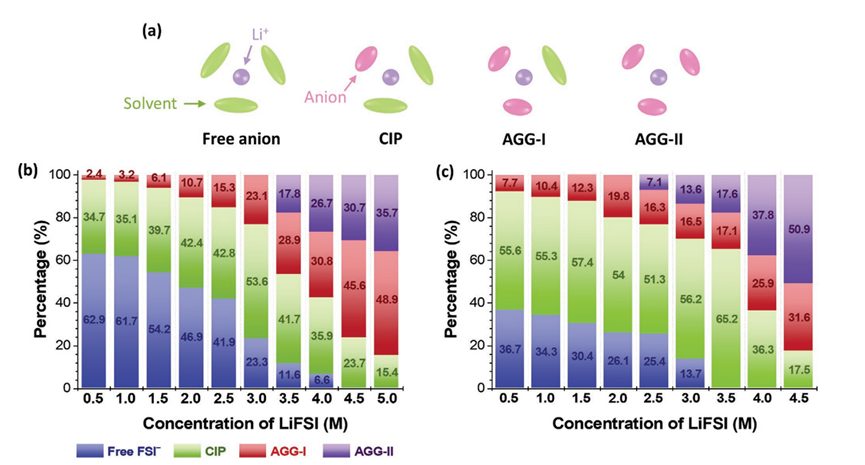
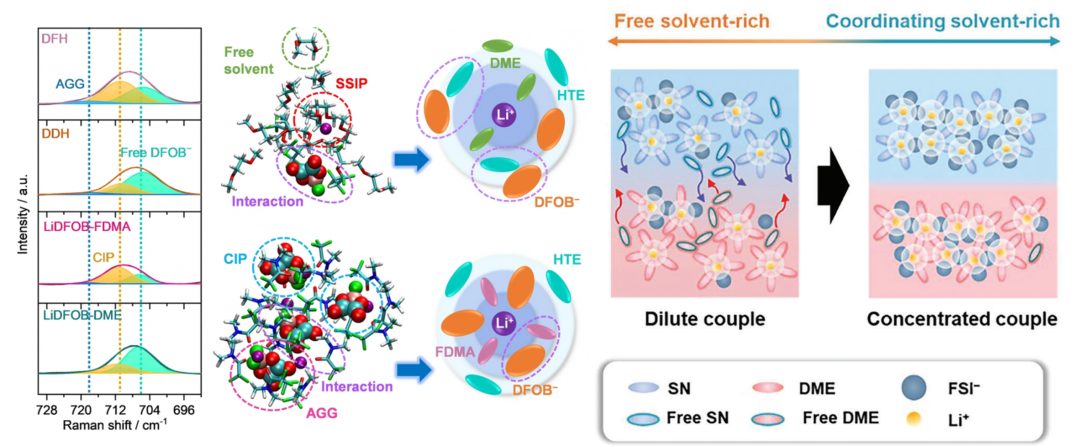
✅8大电解液分析脚本:Li-O配位数 | Li-分子配位数 | SSIPs+CIPs+AGGs | 质心+离子运动轨迹绘制 | 溶剂分子区分+命名 | 自由溶剂占比
✅计算表格+Origin制图模板+结构出图美化:锂盐与溶剂分子配比、RDF+配位数双纵轴图、配位占比饼状图、离子对占比组合柱状图、溶剂化结构图、HOMO/LUMO/静电势图美化、运动轨迹图、离子迁移孔道图
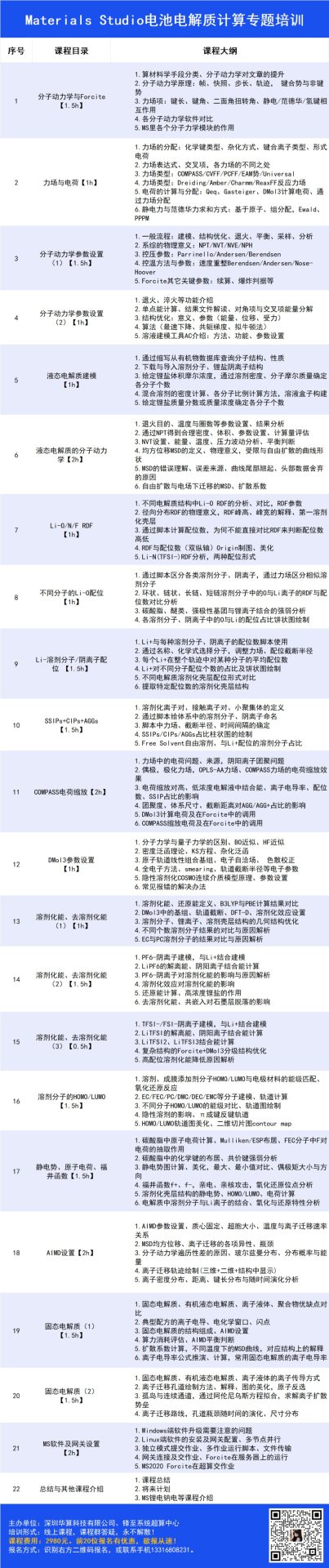

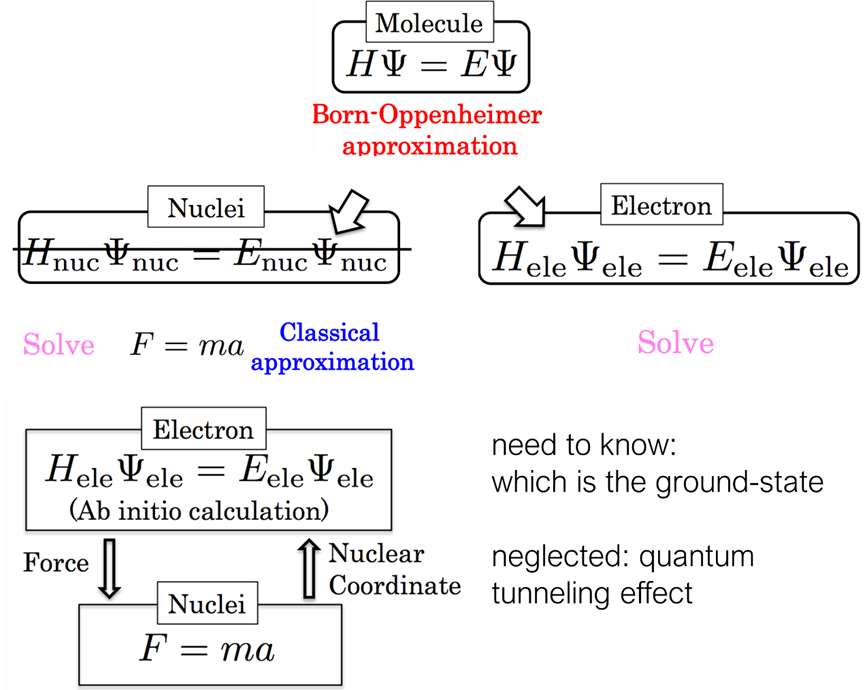
1. 计算材料学手段分类、分子动力学对文章的提升
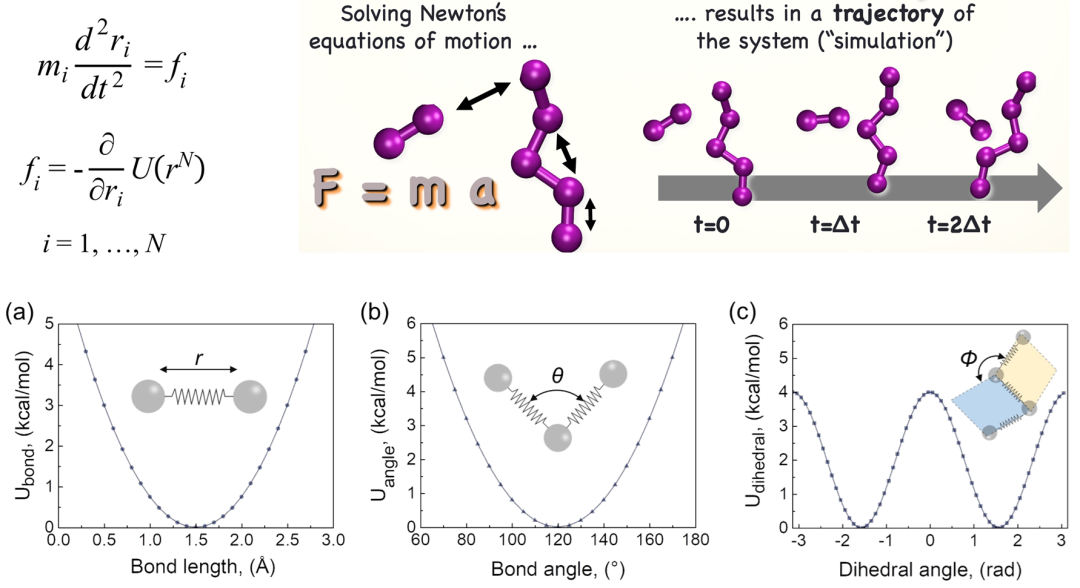
2. 分子动力学原理:帧、快照、步长、轨迹, 键合势与非键势
3. 力场项:键长、键角、二面角扭转角、静电/范德华/氢键相互作用
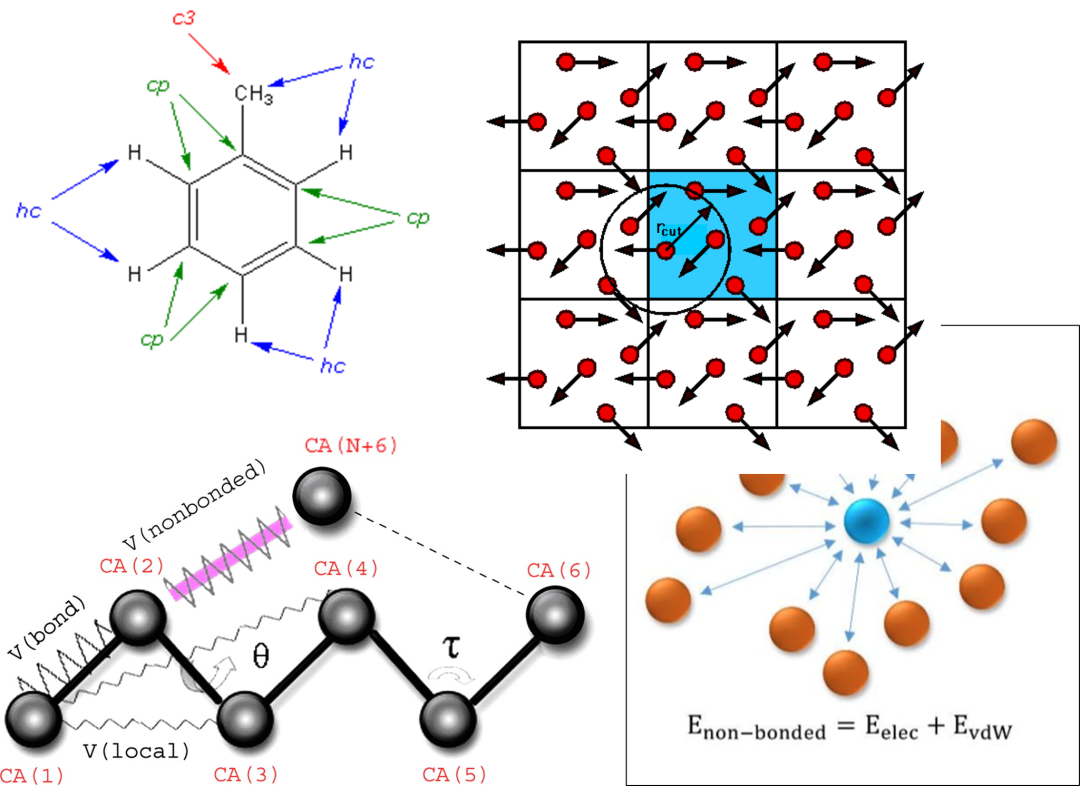
1. 力场的分配:化学键类型、杂化方式、键合离子类型、形式电荷
3. 力场类型:COMPASS/CVFF/PCFF/EAM势/Universal等
4. 力场类型:Dreiding/Amber/Charmm/ReaxFF反应力场
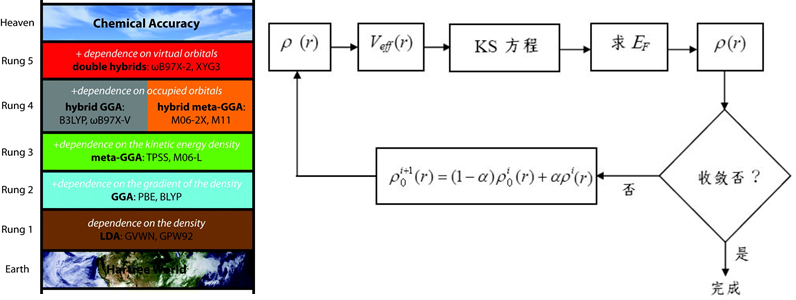
5. 电荷的计算与分配:Qeq, Gasteiger,DMol3计算电荷,通过力场分配
6. 静电力与范德华力求和方式:基于原子、组,Ewald、PPPM
1. 一般流程:建模、结构优化、退火、平衡、采样、分析
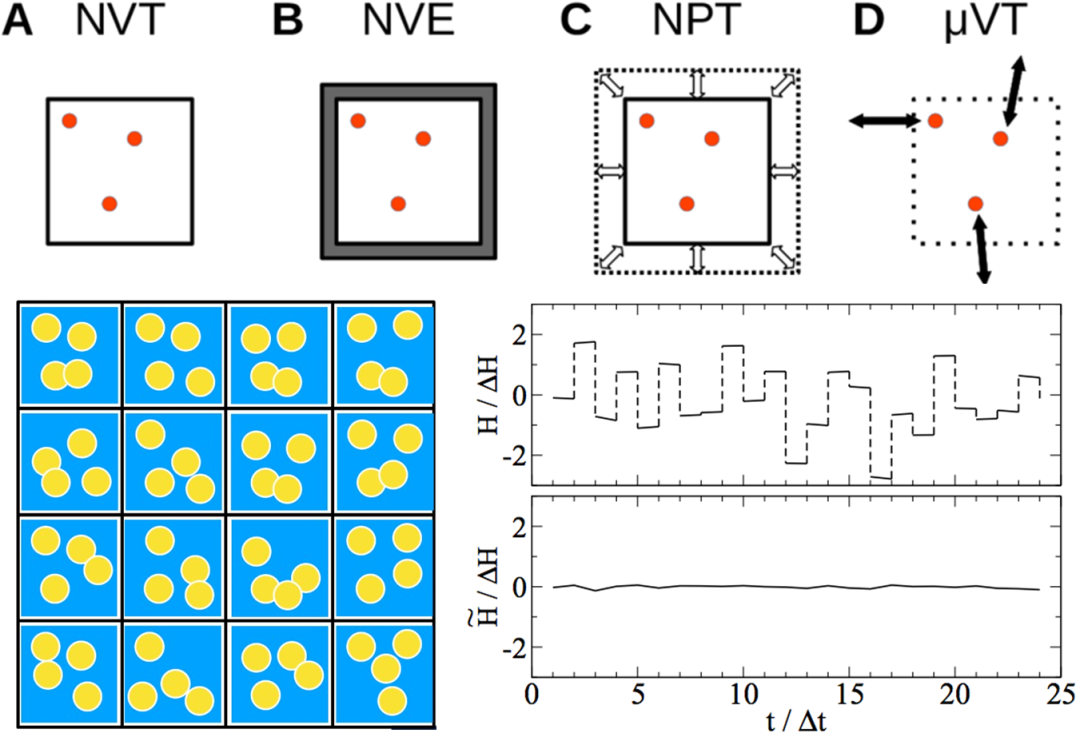
2. 系综的物理意义:NPT/NVT/NVE/NPH
3. 控压参数:Parrinello/Andersen/Berendsen
4. 控温方法与参数:速度重整/Berendsen/Andersen/Nose-Hoover
5. Forcite其它关键参数:续算、爆炸判据等
1. 退火、淬火等功能介绍
2. 单点能计算、结果文件解读、对角项与交叉项能量分解
3. 结构优化:意义、参数
4. 算法(最速下降、共轭梯度、拟牛顿法)
5. 电解液建模工具AC介绍:方法、功能、参数设置
3. 给定锂盐体积摩尔浓度,通过溶剂密度、分子摩尔质量确定各分子个数
4. 混合溶剂的密度计算、各分子比例计算方法,溶液盒子构建
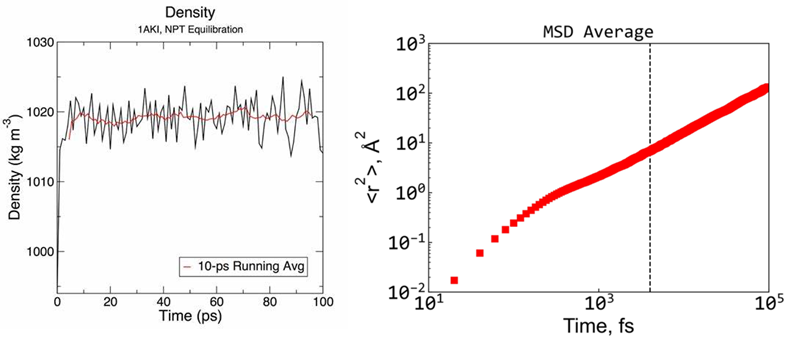
2. 通过NPT得到合理密度、体积、参数设置、计算量评估
3. NVT设置、能量、温度、压力波动分析、平衡判断
4. 均方位移MSD的定义、物理意义,受限与自由扩散的曲线形状
5. MSD的错误理解、误差来源、曲线尾部翘起、头部数据舍弃的原因
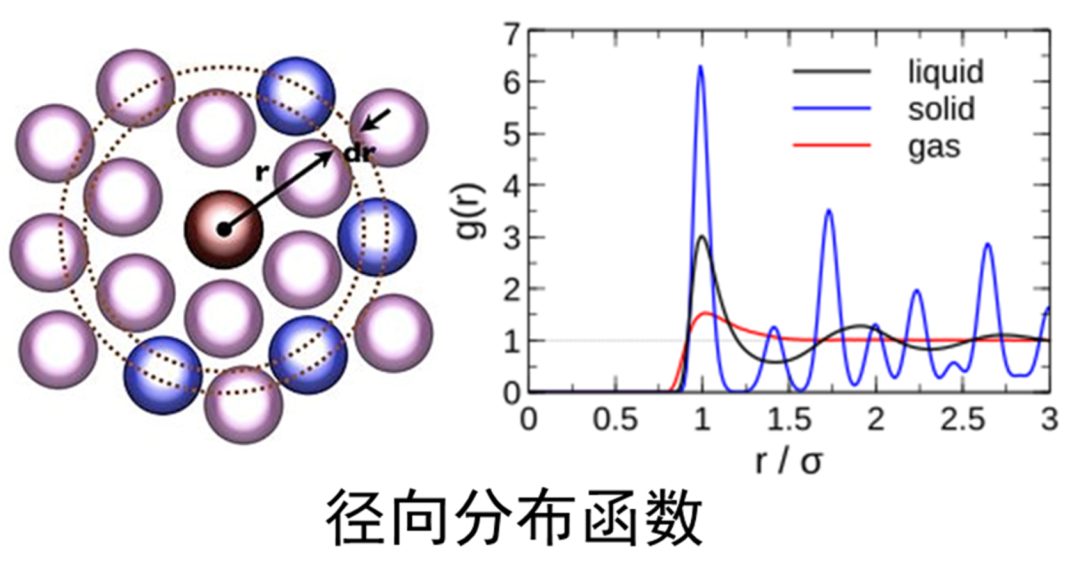
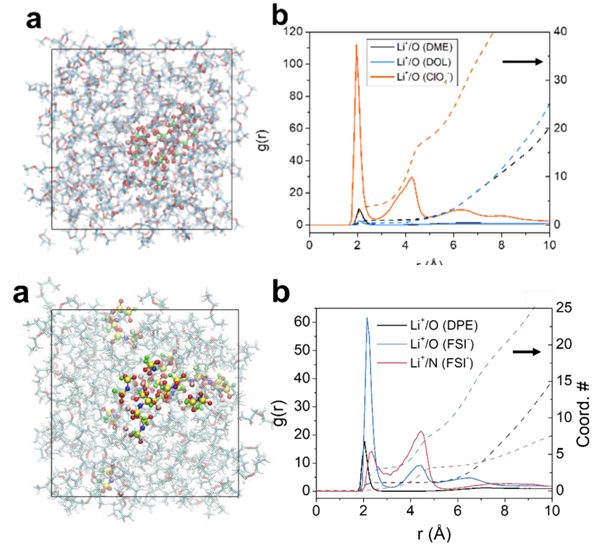
1. 不同电解质结构中Li-O RDF的分析、对比,RDF参数
2. 径向分布RDF的物理意义,RDF峰高、峰宽的解释、第一溶剂化壳层
3. 通过脚本计算配位数,为何不能直接对比RDF来判断配位数高低
4. RDF与配位数(双纵轴)Origin制图、美化
5. Li-N(TFSI–)RDF分析,两种配位形式
1. 通过脚本区分各类溶剂分子、阴离子,通过力场区分相似溶剂分子
2. 环状、链状、长链、短链溶剂分子中的O与Li离子的RDF与配位数对比分析
3. 碳酸脂、醚类、强极性基团与锂离子结合的强弱分析
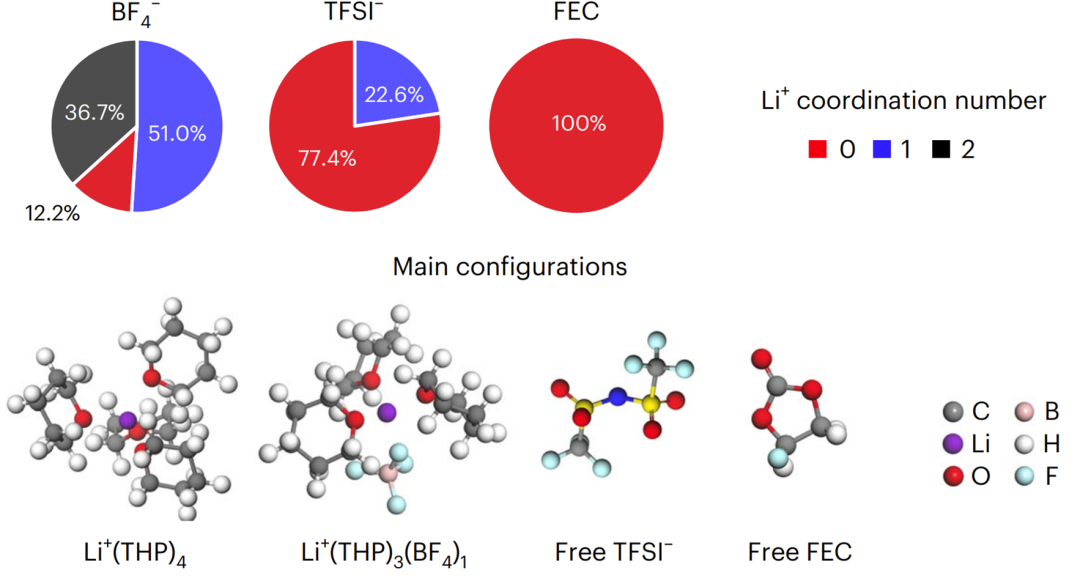
4. 各溶剂分子、阴离子中的O与Li的配位占比饼状图绘制
1. Li+与每种溶剂分子、阴离子的配位数脚本使用
2. 通过名称、化学式选择分子,调整力场、配位截断半径
3. 每个Li+在整个轨迹中对某种分子的平均配位数
4. SSIPs/CIPs/AGGs占比柱状图的绘制
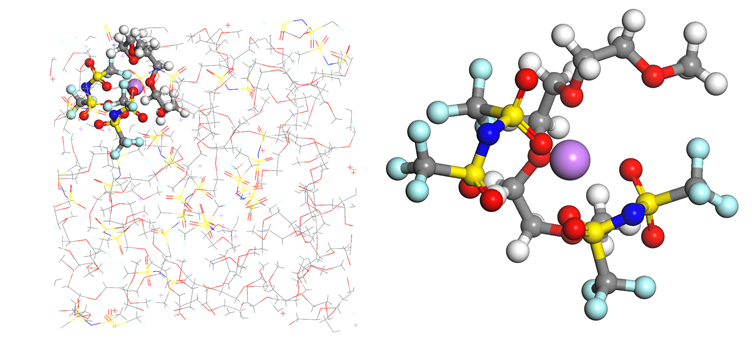
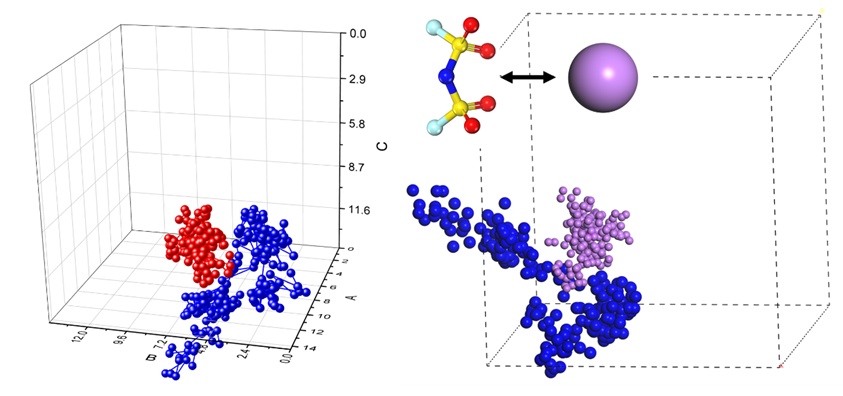
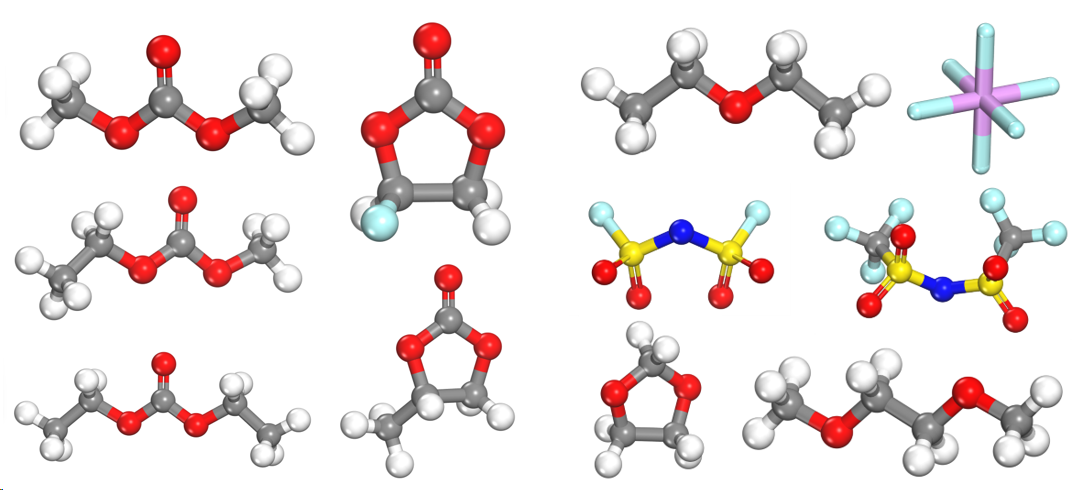
1. FEMC/FEC/D2/FSI–分子结构建模,溶液盒子建模+平衡
2. FSI–从第二溶剂壳层靠近Li+并结合的动态过程
4. 通过运动轨迹观察Li+与FSI–结合的动态过程
3. 原子轨道线性组合基组、电子自洽场、 色散校正
4. 全电子方法、Smearing、轨道截断半径等电子参数
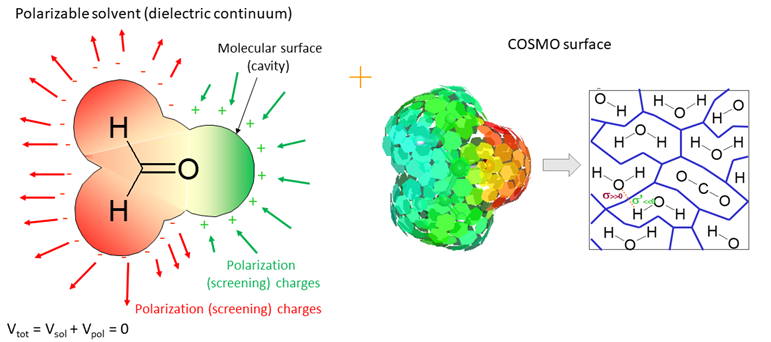
5. 隐性溶剂化COSMO连续介质模型原理、参数设置
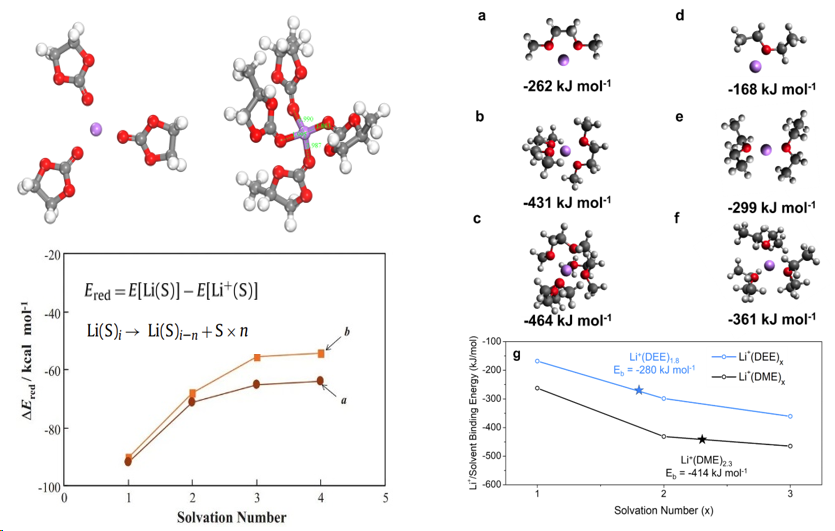
1. 溶剂化能、还原能定义、B3LYP与PBE计算结果对比
2. DMol3中的基组、轨道截断、DFT-D、溶剂化效应设置
3. 溶剂分子、锂离子、溶剂壳层结构的几何结构优化
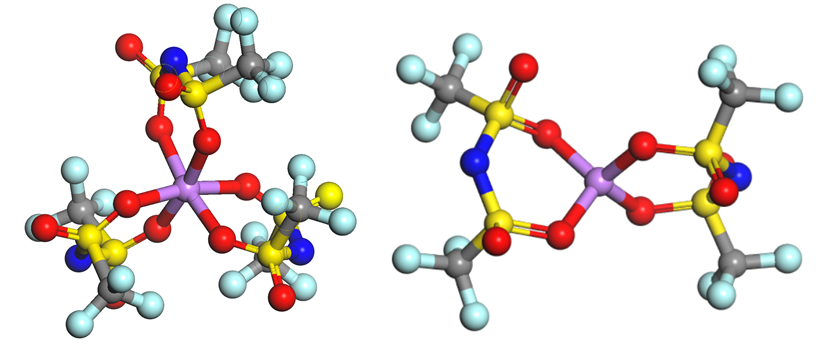
1.TFSI–/FSI–阴离子建模,与Li+结合建模
2. LiTFSI的解离能、阴阳离子结合能计算
3. LiTFSI2、LiTFSI3结合能计算
4. 复杂结构的Forcite+DMol3分级结构优化
5.高配位溶剂化能降低原因解析
-
溶剂、成膜添加剂分子HOMO/LUMO与电极材料的能级匹配、氧化还原反应
-
EC/FEC/PC/DMC/DEC/EMC等分子建模、轨道计算
-
-
-
HOMO/LUMO轨道图美化、二维切片图contour map
1. 碳酸脂中原子电荷计算、Mulliken/ESP布居、FEC分子中F对电荷的抽取作用
3. 静电势图计算、美化,最大、最小值对比、偶极矩大小与方向
4. 福井函数f+、f–,亲电、亲核攻击,氧化还原位点分析
5. 溶剂化壳层结构的静电势、HOMO/LUMO、电荷计算
6. 电解质中溶剂分子与Li离子的结合、氧化与还原特性分析
1. AIMD参数设置、质心固定、超胞大小、温度与离子迁移速率关系
3. 分子动力学遍历性差的原因、玻尔兹曼分布、分布概率与能量
5. 离子密度分布、距离、键长分布与随时间演化分析
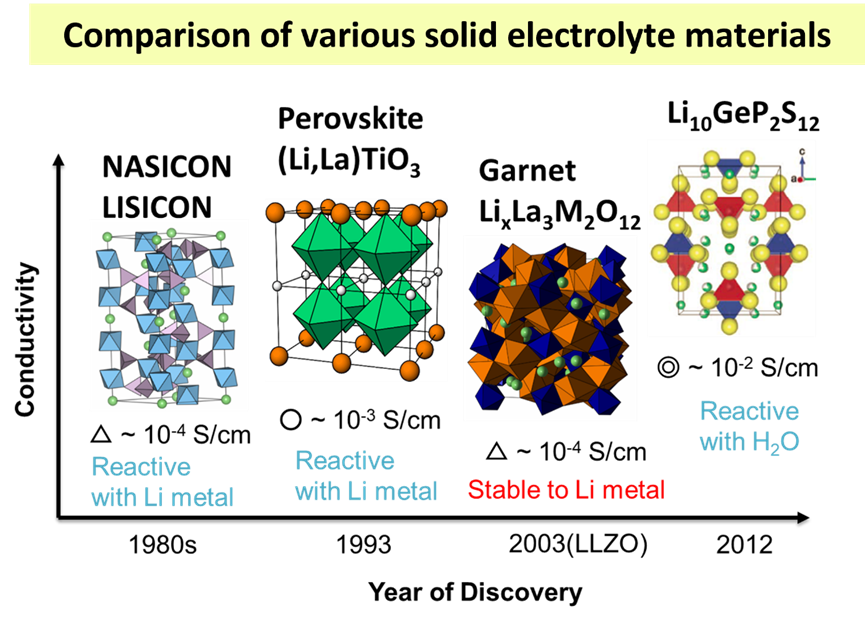
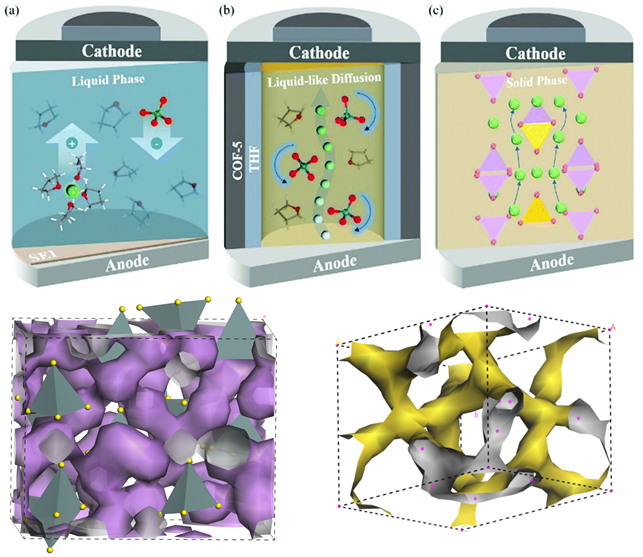
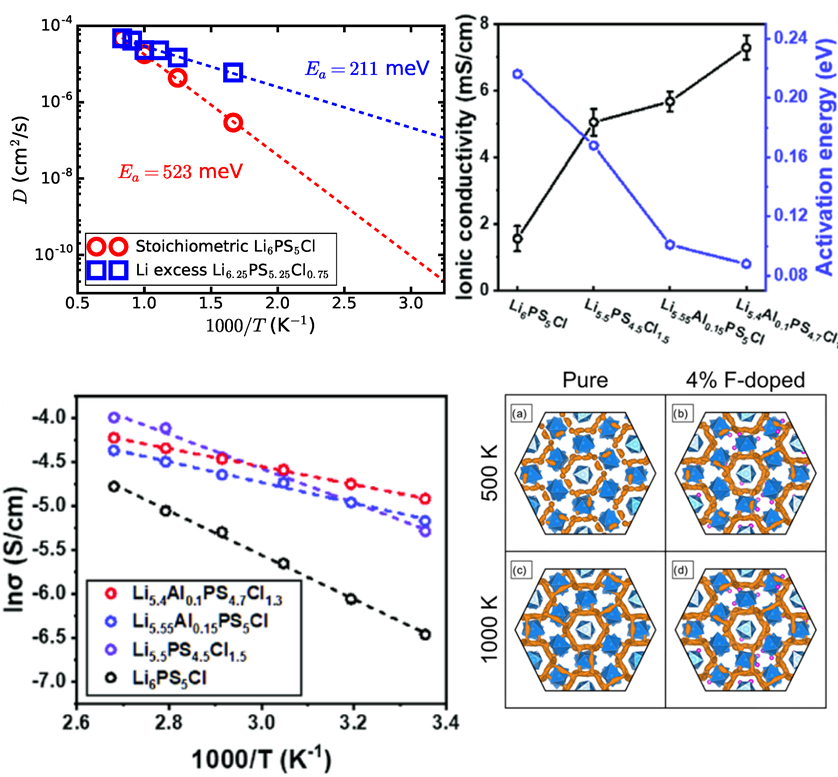
1. 固态电解质、有机液态电解质、离子液体、聚合物优缺点对比
5. 扩散系数计算,不同温度下的MSD曲线,对应结构上的解释
6. 离子电导率公式推演、计算,常用固态电解质的离子电导率
1. 固态电解质、有机液态电解质、离子液体的离子传导方
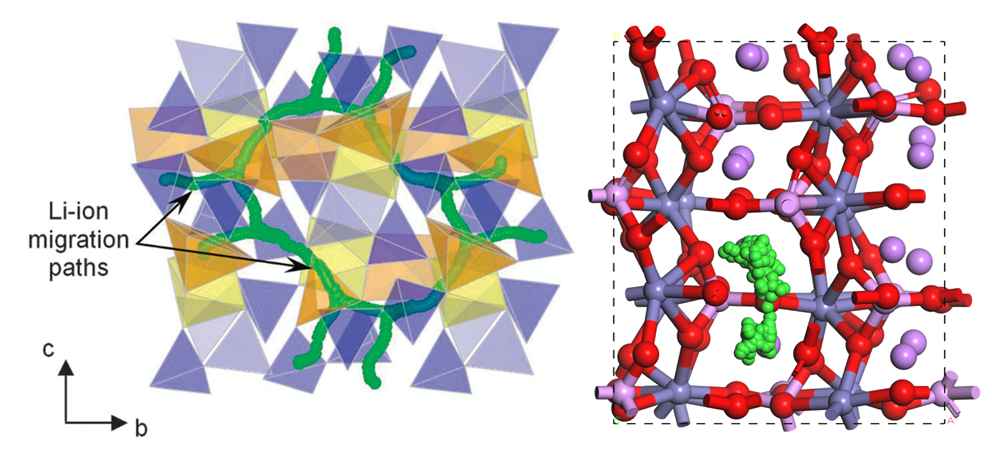
2. 离子迁移孔道绘制方法、解释、图的美化,原子反选
3. 孤岛与连续通道,通过阿伦尼乌斯方程拟合,求解离子扩散势垒
4. 离子迁移路线,孔道瓶颈随时间的演化、尺寸分布
2. Linux端软件的安装及网关配置、多节点并行
4. 网关连接及交作业、Forcite在服务器上的运行
主办单位:深圳华算科技有限公司(拥有VASP、Materials Studio、Gaussian、LAMMPS商业版权)
深圳浦华系统技术有限公司(Materials Studio官方代理)
培训形式:线上课程,可无限次观看,课程群永不解散,随时提问,及时解答。
课程费用:2980元。名额有限,欲报从速!提供增值税普通发票及邀请函。与MS电池计算专题课、MS光电热催化计算专题培训、MS半导体计算专题培训同时报名优惠更多哦!
报名方式:识别下方二维码报名,或者联系手机133-1680-8231。