



电化学O2活化为单线态氧(1O2)的高效合成提供了一种绿色途径。然而,它通常是由吸附依赖的O2活化和转化决定的,并且可能受到表面结合的*OOH的缓慢解吸的影响。
上海交通大学张礼知教授、么艳彩副教授等人报道了一种不依赖于吸附的O2活化途径,该途径通过压缩应变金红石型TiO2(CSR-TiO2)的O2单氢工艺电合成1O2。该CSR-TiO2的O2生成速率为148.26 μmol l-1 min-1,法拉第效率接近100%,优于无应变的对应物(35.97 μmol l-1 min-1)和其他先前报道的材料。CSR-TiO2的优异性能源于压缩应变,压缩应变可以抑制O2吸附的还原不饱和位点的形成,增强原子氢(H*)的还原能力,有利于O2的单加氢途径,绕过传统的表面结合*OOH的脱附途径。生成的1O2可用于硫代苯甲醚及其衍生物的选择性氧化,为绿色有机合成提供了一种很有前途的策略。
相关工作以《Compressive-strained rutile TiO2 enables O2 mono-hydrogenation for singlet oxygen electrosynthesis》为题在《Nature Synthesis》上发表论文。第一作者:王瑞兆(上海交通大学2022级硕士研究生)。
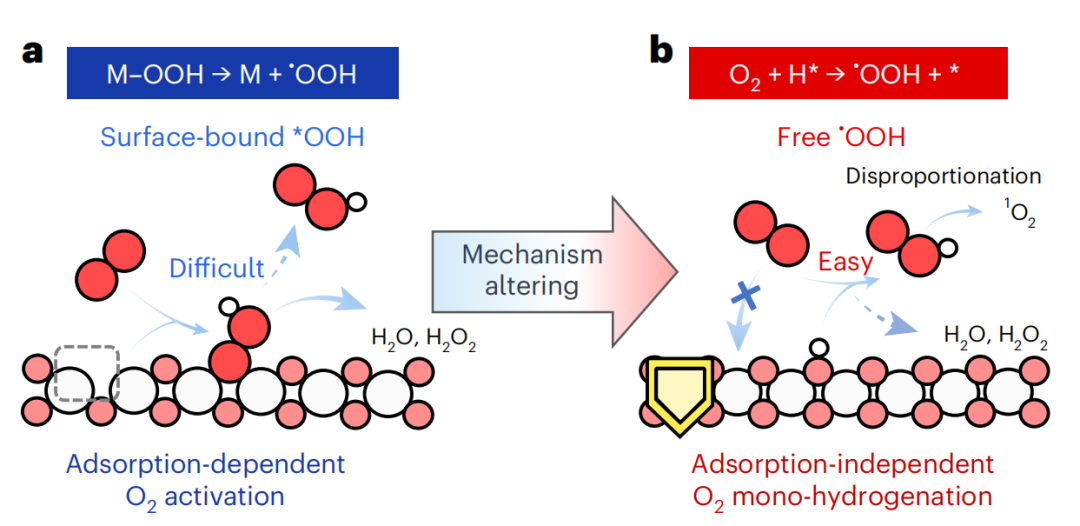
图1 电合成1O2的策略
*O2和*OOH在电催化剂上相似的结合模式导致了它们的吸附和解吸之间的结垢关系,其中强的金属-氧(M-O)结合有利于O2的吸附和活化,但不利于*OOH的解吸(图1a)。尽管经过了广泛的努力,但实现高效、高选择性的1氧合成仍然是一个挑战。
一般来说,•OOH自由基可以在第三者的辅助下,通过高活性H供体如HX(其中X为Cl、Br或I)和氢原子H与O2的吸氢反应自发形成。同时,在电化学体系中,通过经典的Volmer步骤,单电子O2还原为•OOH/•O2–涉及到用一个电子还原H+以产生高活性的氢(H*),然后触发随后的O2单氢化过程,形成自由的•OOH。设计具有较强的H*生成和存储能力以及较差的O2吸附能力的电催化剂,有利于O2单氢途径产生游离的•OOH,而不需要传统的表面结合*OOH解吸过程,从而实现高效的1O2电合成(图1b)。
本文采用晶格约束的方法在TiO上构建了一种设计良好的压缩应变金红石型TiO2(CSR-TiO2)层,旨在通过O2单氢化途径实现优异的电化学合成1O2。通过综合实验和理论计算,阐明了压缩应变对CSR-TiO2中Ti与晶格O相互作用的增强,以及压缩应变对电化学O2活化和自由•OOH/•O2–生成的影响。
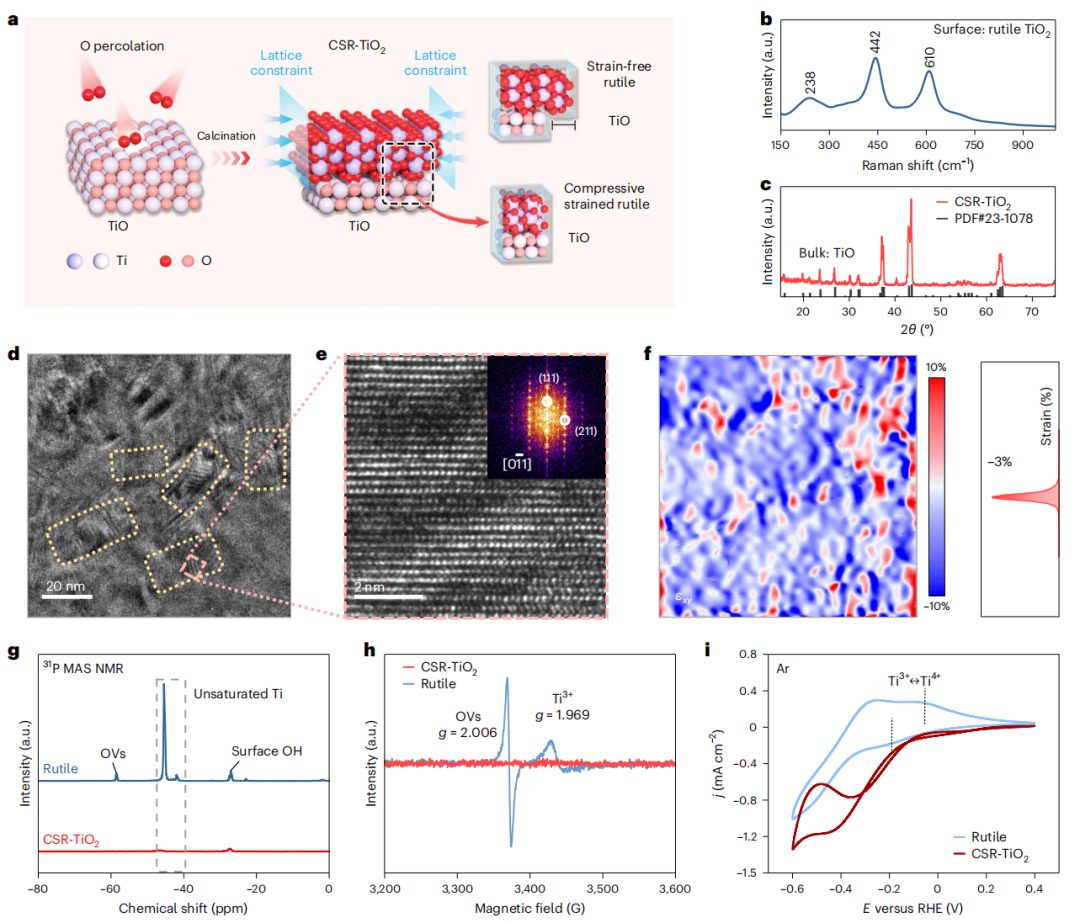
图2 CSR-TiO2的合成与表征
在空气热处理下,通过晶格约束原子重排在一氧化钛(TiO)表面构建CSR-TiO2(图2a)。TiO2表面很容易被氧化成每个Ti原子体积更大的富氧TiO2,而较小的TiO晶胞极大地约束了新形成的TiO2的晶格,从而引发TiO2中面内压应力的形成。拉曼光谱显示新形成的TiO2层在238、442和610 cm-1处出现明显的信号峰,对应于金红石型TiO2(图2b)。XRD分析证实了在TiO表面覆盖了CSR-TiO2层,这可以阻止O原子进一步向TiO内部扩散(图2c)。通过TEM图像(图2d)和AC-HRTEM图像(图2e)观察到的密集皱褶和轻微畸变的单晶反映了CSR-TiO2的表面压应力,其特征是(111)和(211)晶面对应的面间距分别为2.17 Å和1.67 Å。
因此,CSR-TiO2主要以双轴压缩应变的(0-11)晶面暴露,而TiO则以密集排列的Ti-O骨架的(001)晶面暴露,表明在相同Ti密度下,CSR-TiO2中TiO与TiO2的晶格参数差异约为14%。这种巨大的晶格差异极大地限制了TiO2晶格的膨胀,导致CSR-TiO2表面出现压缩应变,甚至可见褶皱(图2e)。随后,采用几何相位分析(GPA)定量评估了CSR-TiO2表面的压应变程度,发现CSR-TiO2在水平方向上表现出约3%的压应变(图2f)。然后用三甲基膦(P(CH3)3)、低温固态电子顺磁共振(EPR)表征催化剂表面的不饱和位点(图2g、h)。在CSR-TiO2中只观察到与表面-OH基团(-28 ppm)相关的信号,排除了其表面存在还原性缺陷位点,如OV和Ti3+位点。
对电化学还原过程中可能产生的还原性缺陷进行了分析。如图2i所示,金红石在Ar气氛下的CV扫描显示,在0 V附近有一对明显的氧化还原峰,对应于Ti3+和Ti4+之间的氧化还原,而在CSR-TiO2中没有出现。这一差异表明,CSR-TiO2对负电位的耐受性更强,对生成还原性不饱和位点的电化学还原反应不敏感。
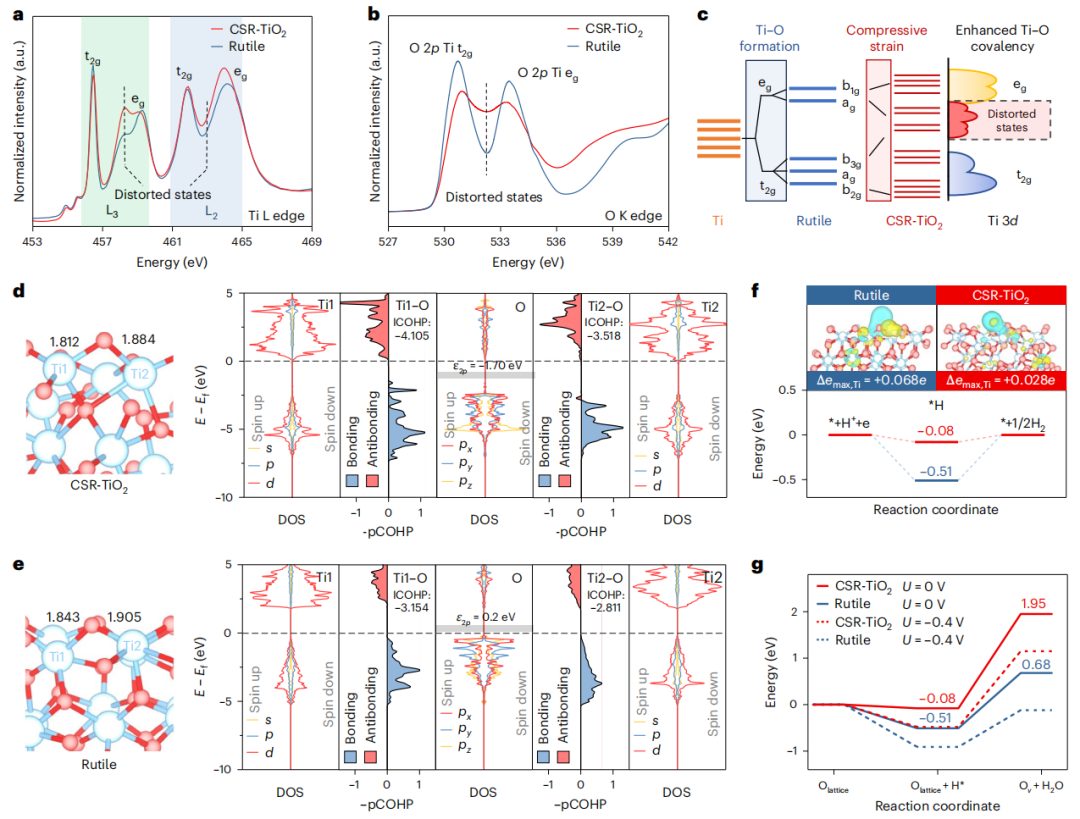
图3 压缩应变增强Ti-O相互作用
图3a中Ti-L2的sXAS谱只有两条线对应于从2p核能级到3d态t2g和eg轨道的跃迁,而Ti-L3边缘则有三个主峰和两个弱前峰。在金红石型TiO2中,Ti-L3边缘的eg相关峰下的肩部在高局部八面体对称性中失去了Ti4+的谱重,而在CSR-TiO2中获得了谱重,表明3d变形增强,这是由压缩应变TiO2引起的。由于O 2p态和Ti 3d态之间的共价混合,在O-K边缘XAS的前边缘区域也可以发现类似的情况。图3b显示金红石的O-K XAS中有两个尖锐的前边缘峰,在CSR-TiO2的情况下,这两个前边缘峰变得很差,表明t2g和eg轨道进一步分裂,如图3c的3d分裂示意图所示。
随后,进行第一性原理计算,进一步研究压缩应变对CSR-TiO2表面暴露的Ti和O与金红石之间的轨道相互作用的影响(图3d、e)。根据CSR-TiO2和金红石表面Ti-O键的pCOHP, CSR-TiO2表面的附加键/反键能级与sXAS对表面电子态变化的实验结果一致。压缩应变增加了CSR-TiO2中Ti-O键的成键轨道和反键轨道的pCOHP,表明压缩应变对Ti-O相互作用的显著增强。同时,CSR-TiO2中O原子的pDOS表明,压缩应变降低了O原子的p轨道能级,从而提高了H*的吸附能,从而提高了限制在CSR-TiO2上的H*的还原能力,促进了随后的O2单氢反应。
然后,分析了CSR-TiO2和金红石的电化学不饱和位点形成过程,包括通过Volmer步骤吸附H形成*OH中间体和*OH离开生成OV和H2O。首先,计算了H吸附后CSR-TiO2和金红石的电子结构变化(图3f)。差分电荷结果表明,H吸附后CSR-TiO2上的电荷分布主要集中在O原子上,而不是Ti原子上,从而阻止了Ti原子积累过多的负电荷形成Ti3+。另一方面,吸附在金红石表面的H注入的电荷被定位在相邻的Ti原子上。随后,计算了整个电化学还原过程所需的能量(图3g),发现不饱和位点的形成能量从金红石表面的0.68 eV增加到CSR-TiO2表面的1.95 eV,证实了CSR-TiO2表面不饱和位点的形成更加困难。

图4 电合成1O2的性能
在单个电解池中使用三电极系统,在连续泵送空气作为O2源的情况下,评估了CSR-TiO2和金红石的1O2电合成性能。首先,利用EPR技术在-0.4 V下检测1O2和其他可能的反应物质。利用TEMP作为探针捕获电化学O2活化过程中产生的1O2(图4、b),在CSR-TiO2电化学体系中出现了较强的TEMPO信号,表明产生了丰富的1O2。相反,在金红石对应物中没有观察到这种TEMPO信号,这表明CSR-TiO2对电化学O2活化具有优越的1O2选择性。在随后的猝灭实验中(图4b),在连续泵送Ar或向CSR-TiO2电化学体系中加入超氧化物歧化酶后,TEMPO信号消失,表明其1O2电合成对O2供应和•OOH生成有很强的依赖性。
进一步使用ABDA作为荧光探针检测1O2的生成(图4c),发现在CSR-TiO2体系中,ABDA的荧光信号迅速下降,并在10 min内达到最小值,而在金红石体系中,ABDA的荧光信号缓慢下降,1O2生成速率较慢。DMPO探针显示,在CSR-TiO2体系中存在•OOH,并具有显著的DMPO-OOH信号(图4d),而在金红石体系中不存在•OOH。在严格的O2饱和条件下,利用RRDE检测O2还原反应的电子转移数。利用K-L方程拟合,如图4e所示,CSR-TiO2的电子转移数接近1,与O2单电子氢化过程吻合较好。CSR-TiO2的•OOH氧化电流强于金红石,其环电位选择为•OOH选择性氧化(图4f)。
本文采用糠醇(FFA)探针降解实验,定量评价连续气流下CSR-TiO2、TiO和金红石的1O2生成速率。如图4所示,CSR-TiO2的降解速率最高,为0.0813 min-1,是金红石的2.4倍。通过排除FFA在阳极上的直接氧化,得到了每种催化剂的1O2电合成速率(图4h)。与已有的1O2生成系统相比,基于CSR-TiO2的电化学O2活化体系在1O2生成方面具有优势(图4i)。
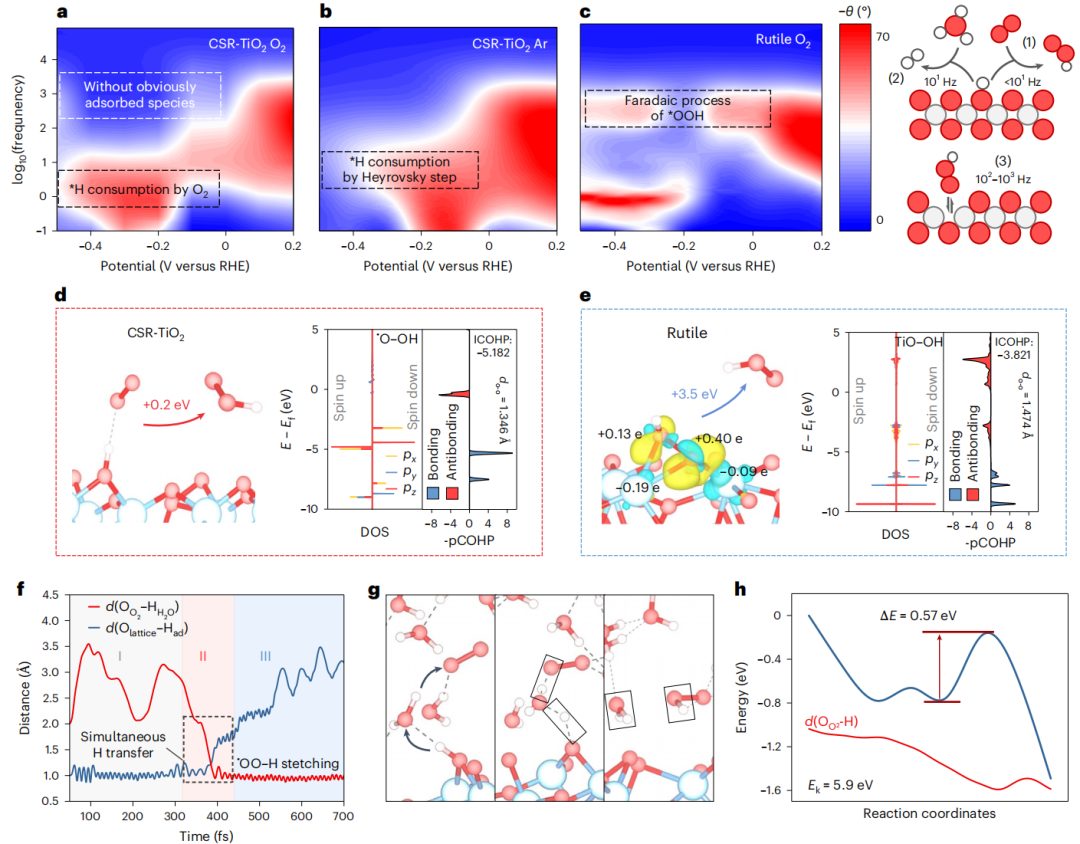
图5 机理研究
本研究利用电化学阻抗谱(EIS)监测了操作条件下CSR-TiO2和金红石的O2活化过程。如图5a-e所示,CSR-TiO2和金红石的波德图表现出不同的相角分布。在CSR-TiO2的低频区,观察到相位角从Ar气氛下的101 Hz转变为O2气氛下的100 Hz。这一转变表明CSR-TiO2从H+的H*消耗(Heyrovsky步骤)转变为O2的H*消耗(O2单氢化)。通过气相色谱法分析不同气氛下的产物,证明了Heyrovsky步骤与CSR-TiO2表面单氢化之间的竞争关系。金红石在中频范围内表现出明显的相角分布,与*OOH与电极表面之间的电荷转移阻抗响应有关。相比之下,CSR-TiO2在相同频率区域的变化很小,表明不存在*OOH。
从理论上计算了CSR-TiO2上O2单氢化途径和金红石上*OOH均裂途径产生自由•OOH的热力学差异。从热力学角度看,CSR-TiO2表面的单加氢途径仅需0.2 eV即可产生游离•OOH(图5d)。同时,需要3.5 eV的外部能量才能实现M-O键的均裂,在金红石上形成自由的•OOH(图5f)。差分电荷和Bader电荷结果表明,表面结合的*OOH与金红石上的Ti原子之间存在强烈的相互作用,阻碍了*OOH与表面的分离。*OOH的O-O键被拉长和减弱,有利于O-O键的裂解,而不是M-O键的均裂。
为了了解CSR-TiO2表面的O2单加氢过程,在外加偏压的情况下进行了AIMD模拟,以跟踪反应过程。通过引入一个额外的氢原子,在模拟系统中引入了一个恒定的负电位。O2单氢化过程的模拟过程可以根据参与原子之间距离的变化分为三个阶段(图5h):(1) O2的引入和稳定;(2)在水分子与H*形成氢键的帮助下,O2的扩散和旋转到一个合适的构象,以促进单氢化过程,从而形成•OOH(图5i);(3)•OOH离开原来的H*位点。如图5g所示,O2单加氢过程只需要0.57 eV的能垒,低于整个体系的动能(5.9 eV),证实了O2单加氢过程在CSR-TiO2体系中是动态可行的。
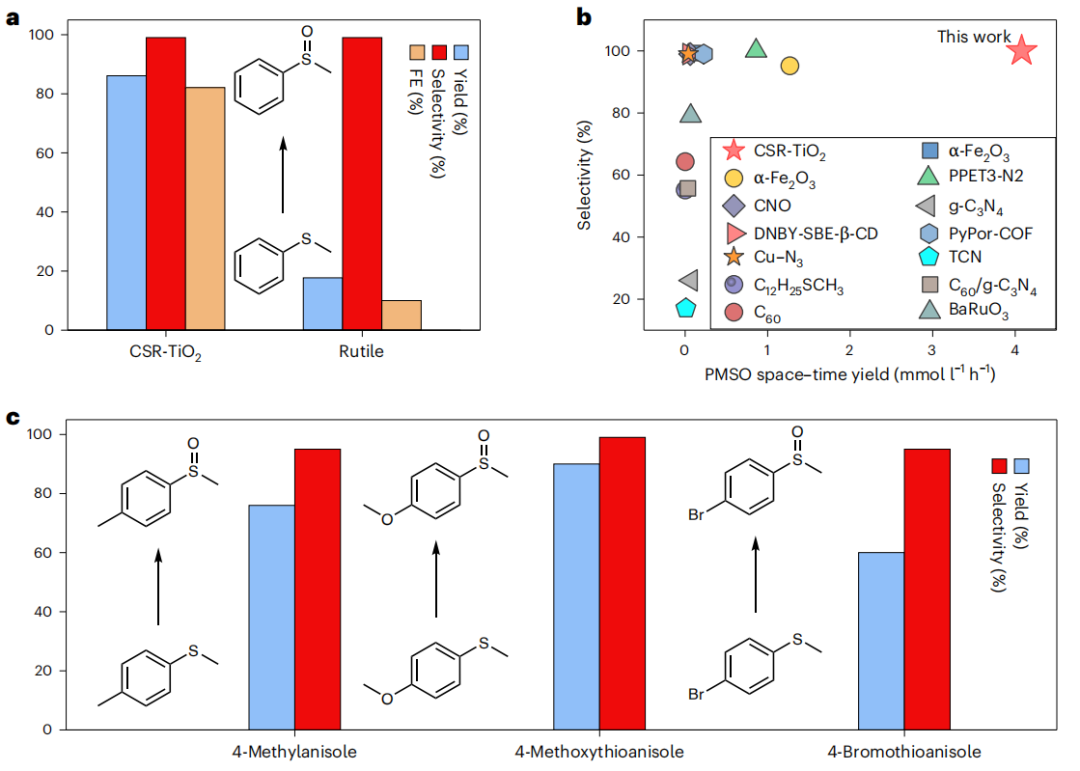
图6 茴香硫醚的选择性亚砜化反应
鉴于该CSR-TiO2电化学生成1O2的高效率和高选择性,以硫苯甲醚选择性亚砜化为例,研究了其在精细化学品合成中快速选择性氧化反应的潜力。在2 h内,苯甲酰亚砜(PMSO)的产率达到86.1%,选择性为99.50%,法拉第效率为82.1%(图6a)。相比之下,金红石体系的PMSO产率仅为17.7%,法拉第效率为10.0%(图6b)。令人印象深刻的是,与之前的报道相比,CSR-TiO2电化学体系表现出压倒性的PMSO转化性能,即使是三种不同的硫代异唑衍生物(图6c)。
Compressive-strained rutile TiO2 enables O2 mono-hydrogenation for singlet oxygen electrosynthesis,Nature Synthesis,2025.
https://www.nature.com/articles/s44160-025-00756-0
👉 点击阅读原文,立即下单!