

说明:本期主要讲述同步辐射HERFD-XAS技术特点及原位HERFD-XAS在电催化领域中的应用!想要了解更多请看历史内容!🥰
👉同步辐射XAFS实验样品的厚度/重量如何确定?选透射还是荧光?SAMPLEM4M一键解决!!
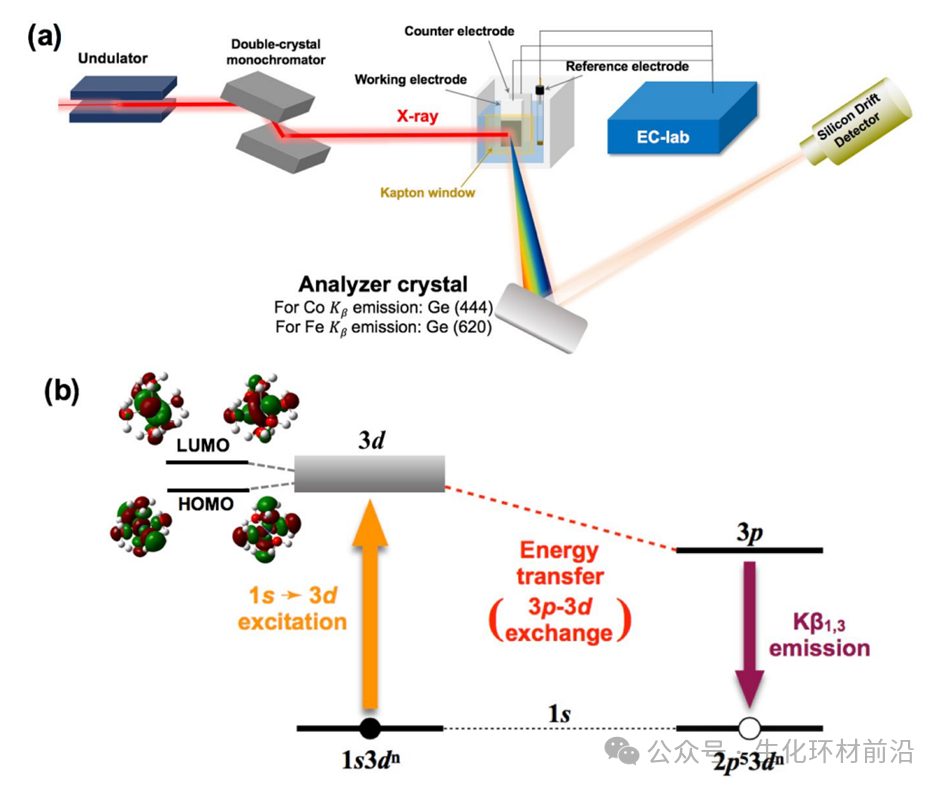

图1应用于液体电化学池的原位HERFD-XAS装置的示意图和HERFD-XAS的能级图
能量分辨率较差的限制因素是核心空穴的固有寿命展宽,而非光源分辨率。其原理是,当入射光子的能量Ω=hυ1将K壳层电子(即1s电子)激发出到未占据态或连续态时,基态电子信息被激发到具有核心空穴的中间态(图1)。外层电子填充空穴后,会产生能量为ω=hυ2的荧光辐射衰减或俄歇电子发射,最终状态为上轨道中的空穴。Heisenberg不确定性原理(Γ~h/Δt)提供了一个与寿命(Δt)成反比的能量不确定性。因此,这一过程中的中间态涉及具有有限寿命的核心空穴,导致发射线的洛伦兹展宽Γ。
Γ(有效寿命宽度)=Γint+Γf in
其中Γint和Γf in分别是中间态和最终态的核心空穴寿命宽度。
注意:核心空穴寿命展宽导致的能量宽度随原子序数增加而指数增长,而L壳层的寿命宽度较K壳层更尖锐,意味着高原子序数元素的X射线吸收光谱能量分辨率较低。
在化学反应中,过渡金属与反应物或中间体形成的化学键主要涉及活性中心的杂化d轨道,这些轨道与吸收位点的局部对称性紧密相关,从而使得XANES的边前区域能够反映位点与吸附物之间的对称性相互作用。由于K边跃迁中1s到(n-1)d态的跃迁是禁阻的,这导致过渡金属中的杂化d态在XANES中的特征非常微弱。加之内在寿命展宽限制了能量分辨率,使得这些边前特征在传统XAS中难以被清晰识别。
HERFD-XAS技术通过高能量分辨率荧光检测,能够提高XANES特征的能分辨率。与传统XAS相比,HERFD-XAS能够补充识别禁阻的1s到(n-1)d跃迁中的杂化d态,提供关于催化剂–吸附物相互作用、对称性和金属–配体电荷转移的深入见解。
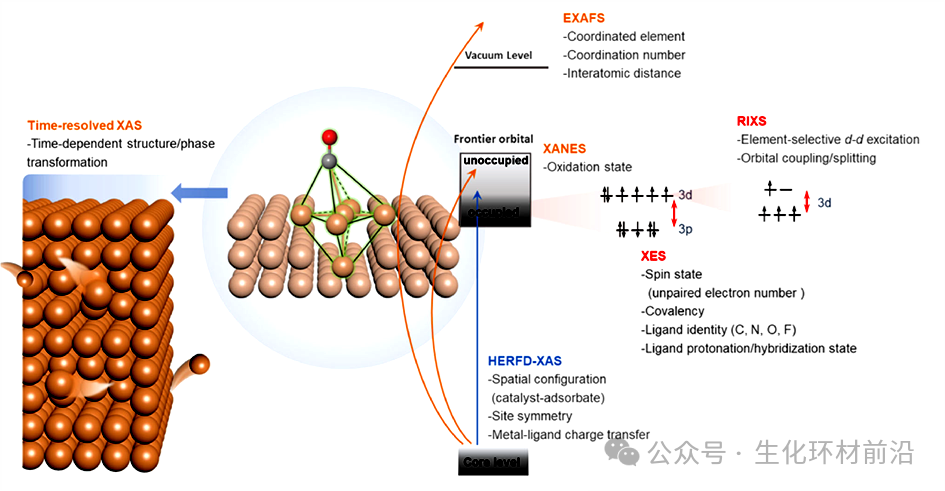
图2 X射线光谱学在解析固液界面电催化中的作用
1️⃣高能量分辨率:HERFD-XAS能够提供比传统XAS更高的能量分辨率,这使得它能够检测到XANES(X射线吸收近边结构)光谱中前缘区域的细微特征,这些特征通常被强烈的吸收边所掩盖。
2️⃣表面敏感性:HERFD-XAS对催化相关的表面具有更高的敏感性,这对于研究催化剂的表面结构和表面反应机制尤为重要。
3️⃣更大的穿透深度:硬X射线的穿透深度比软X射线大, HERFD-XAS能够在不需要高真空条件下进行测量,并且允许使用足够的电解液体积,这对于电化学研究尤其重要。
4️⃣减少背景信号:在特定波长下检测可以显著减少背景信号,与常规XAS相比,这可以提高数据的质量,并允许在其他吸收边缘之外收集数据。
5️⃣适用于Operando条件:HERFD-XAS适用于在工况条件下研究催化剂,尤其在电化学电池中。
6️⃣信息丰富:HERFD-XAS能够提供关于催化剂的详细信息,包括金属–配体相互作用、基底–金属相互作用和金属–吸附物相互作用,这些都是催化过程中的关键因素。
2012年,Lindsay R. Merte 等人[1]利用Operando原位Pt L3边HERFD-XAS来区分质子交换膜燃料电池(PEMFC)阴极上的化学吸附氢、化学吸附氧/羟基和各种铂氧化物。电位依赖的HERFD-XAS光谱的最小二乘拟合结果表明,高电位下观察到的白线强度的增加实际上源于铂氧化物的形成,而不是先前提出的含氧物种的化学吸附。
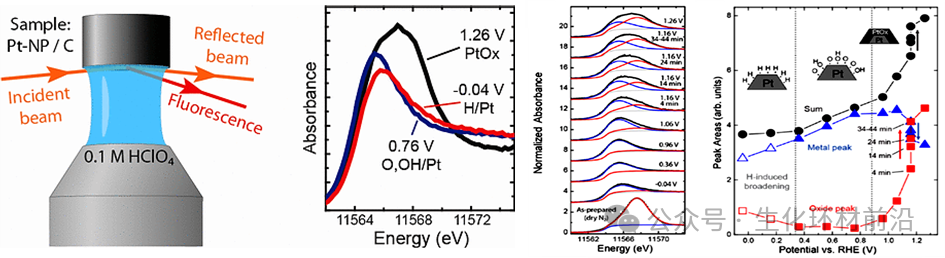
图3玻璃碳上的Pt纳米粒子的原位Pt L3 HERFD-XANES光谱的最小二乘法拟合
与2011年Daniel Friebel[2]的研究报告相比, Pt/Au(111) 与Pt/Rh(111)相比,在1.4 V相对于RHE时的Pt L3 HERFD-XAS光谱的白线强度急剧增加,与同一电位下的Pt溶解相吻合,这意味着Pt/Au(111)中快速氧化物生长是由阳极Pt溶解到Pt2+促进的,随后进一步氧化Pt2+到Pt4+。相比之下,阳极极化将导致Pt/Rh(111)上的钝化,显示出在氧还原反应(ORR)期间减弱的降解过程。
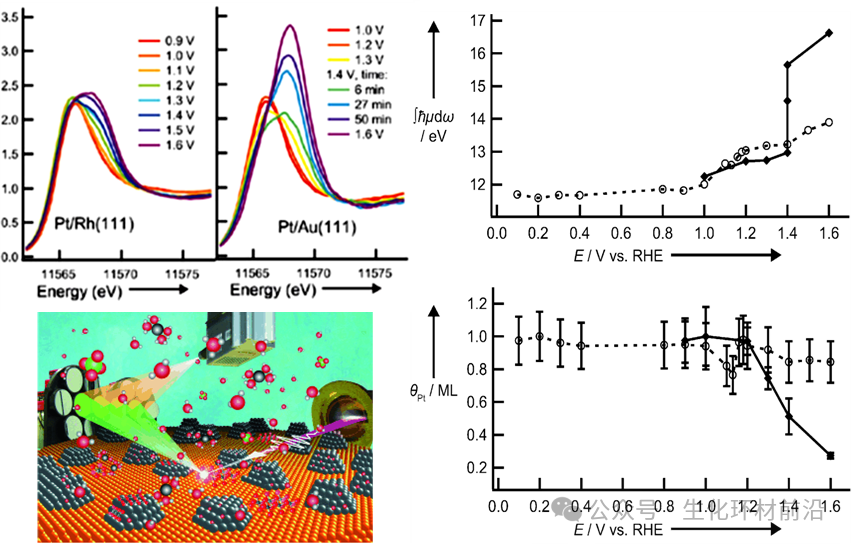
图4 Pt/Rh(111)和Pt/Au(111)在0.01M HClO4中的原位Pt L3 HERFD-XAS光谱
2013年,Daniel Friebel[3]等人利用HERFD-XAS技术确定氧释放反应过程中原位Co电催化剂的化学状态。HERFD-XAS光谱显示随着施加电位的增加,差异几乎不可察觉,这意味着铁离子不参与催化循环。因此,原位HERFD-XAS分析表明Co离子是OER的活性位点,而Fe离子可以有效地稳定Co离子以提供更高的氧化态,从而在OER期间促进Co离子与电解质之间的催化相互作用,导致反应物的稳定中间体,然后是优越的内在OER活性。
HERFD-XAS技术还允许在电催化剂上的反应过程中明确区分表面状态和氧化还原相的变化。例如,Daniel Friebel 利用Operando原位HERFD-XAS方法清晰地追踪Co氧化物/Au(111)在OER期间边前特征的微妙电位诱导变化,这些变化在传统的XAS方式中无法观察到。通过执行最小二乘拟合,最终证明在7710.9 eV处的新特征除了两个CoOOH特征外,可以归因于在高电位下H1-xCoO2相中Co4+离子的存在,这与OER期间主吸收边向高能的移动一致。Co4+的出现被验证对OER性能相当不利。
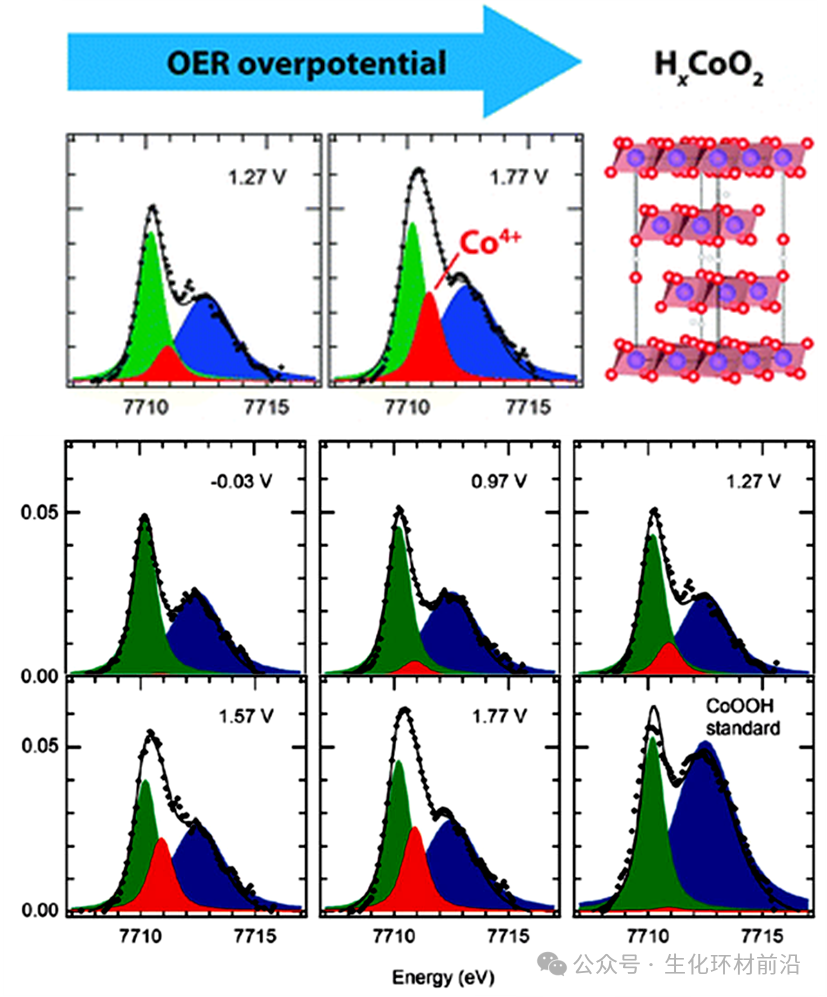
图5钴氧化物/Au(111)的原位Co K边HERFD-XAS前缘特征以及CoOOH的参考
2014年,Daniel Friebe[4]通过Operando原位Cu K边HERFD-XAS光谱具有清晰锐化的特征,证实了在高电位下,单层Cu在Au(111)上直接从金属Cu0相变到CuO,而非在多层Cu/Au(111)中观察到的Cu2O中间体。在低电位下,光谱分析显示,由于Au基底的存在,Cu单层在其金属状态下表现出晶格膨胀(形成Au-Cu合金,然后在其长期运行期间形成Au终止表面),这显著影响CO2电还原反应(CO2RR)中催化表面与中间体之间的相互作用。然而,HERFD-XANES光谱也受到显著的自吸收效应影响,这是在荧光模式下X射线吸收测量中需要考虑的关键因素。对于厚且浓的样品,入射和发射X射线光子的能量依赖性衰减导致光谱特征的失真。因此,为了避免自吸收,应谨慎选择样品浓度以获得准确的HERFD-XAS光谱。
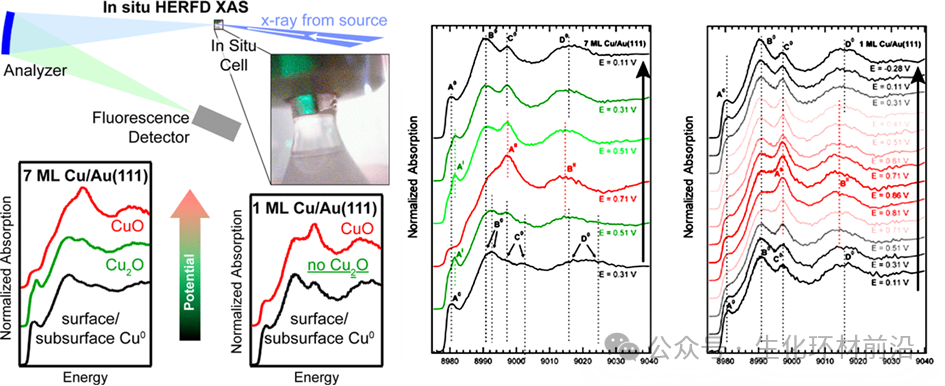
图6不同铜覆盖度f 7ML和g 1ML的Cu/Au(111)的原位Cu K边HERFD-XAS光谱,作为电位的函数
2018年,Hung[5]等人在研究OER过程中,利用原位HERFD-XAS探究二元Co-Fe氧化物中心金属位点的d轨道动态演化。Co K边HERFD-XAS光谱在边前区域显示两个不同区域:(i)低能区域,指的是金属局部四极跃迁;(ii)高能区域,显示氧介导的金属–金属相互作用。对于Co-Dom尖晶石(Fe掺杂的Co氧化物,以Co为主导的晶格框架),HERFD-XAS光谱中的高能位移表明Fe离子显著影响Co离子的氧化行为和/或配位环境。原位Co K边HERFD-XAS光谱显示,随着施加电位的增加,Co-Dom尖晶石样品中关于氧介导的金属–金属相互作用的峰值明显显示出稳定的能量位置和与原始尖晶石相比强度的大幅增加,这证实了Co-Dom尖晶石中氧(2p)和钴(3d)之间强烈的轨道相互作用。
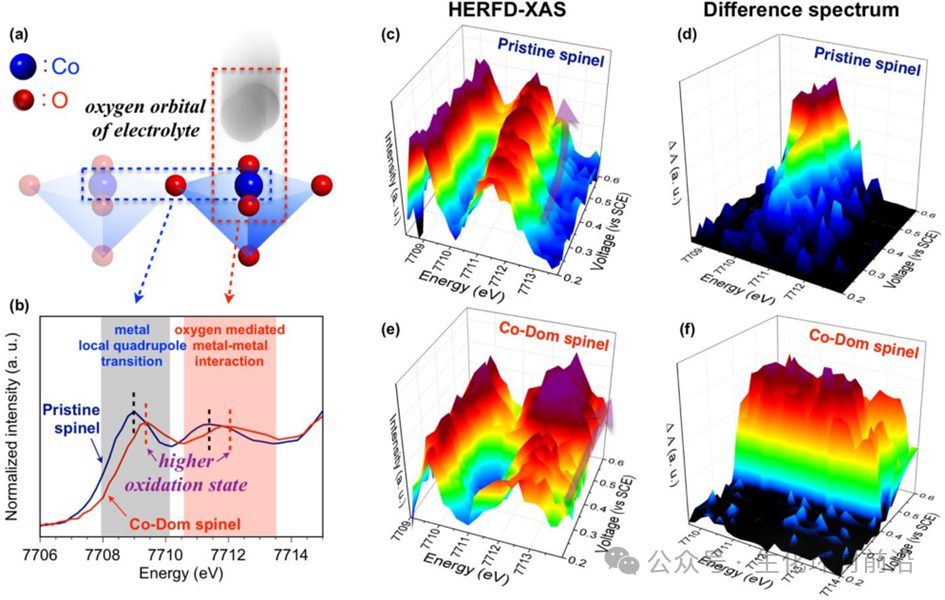
图7各种电化学过程的原位HERFD-XAS分析
2022年,Julian Feijoo[6]等人使用了原位HERFD-XAS在高度活跃的铜纳米催化剂的Operando条件下收集了XANES和EXAFS光谱。研究结果揭示铜纳米催化剂在施加电化学电位后的激活机制,这涉及到表面配体的解离和铜表面的氧化态及配位环境的演变。原始的纳米催化剂最终被还原为具有欠配位活性位点的金属铜纳米颗粒。完全活跃的状态在大约1小时后达到;此时,系统中没有残留的氧化物或配位配体。
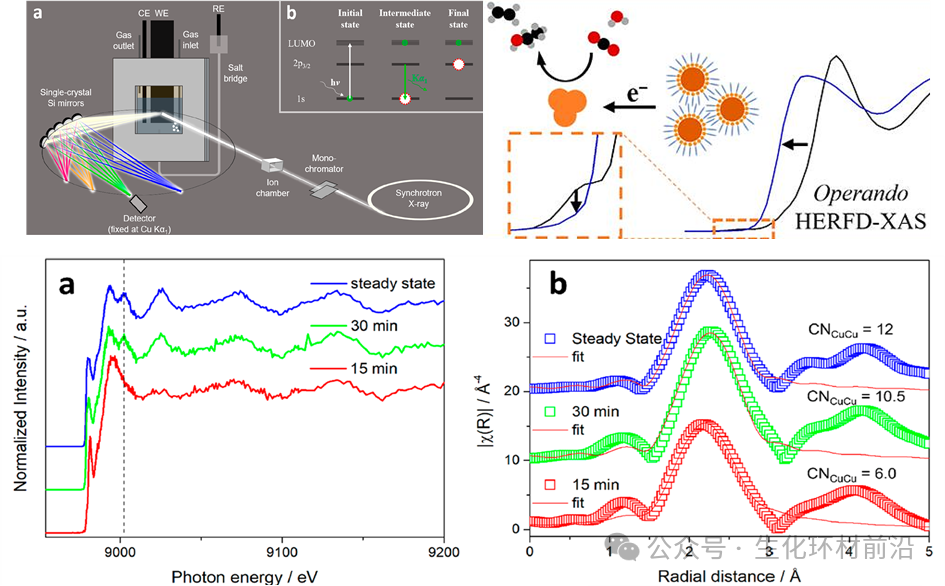
图8Operando条件下收集了XANES和EXAFS光谱
注意:HERFD-XAS主要用于提高XANES光谱的能分辨率,而不是EXAFS光谱。在电化学研究中,由于HERFD-EXAFS需要较长的数据收集时间,因此不适合追踪电化学反应期间的瞬态结构。然而,为了深入研究电催化固液界面,能够在可接受的时间内获得高质量的EXAFS光谱是至关重要的,以便理想地区分光谱特征。
参考文献
[1] Merte Lindsay R., Behafarid Farzad, Miller Daniel J., et al. Electrochemical Oxidation of Size-Selected Pt Nanoparticles Studied Using in Situ High-Energy-Resolution X-ray Absorption Spectroscopy [J]. ACS Catalysis, 2012, 2(11): 2371-2376.
[2] Friebel Daniel, Miller Daniel J., Nordlund Dennis, et al. Degradation of Bimetallic Model Electrocatalysts: An In Situ X-Ray Absorption Spectroscopy Study [J]. Angewandte Chemie International Edition, 2011, 50(43): 10190-10192.
[3] Friebel Daniel, Bajdich Michal, Yeo Boon Siang, et al. On the chemical state of Co oxide electrocatalysts during alkaline water splitting [J]. Physical Chemistry Chemical Physics, 2013, 15(40): 17460-17467.
[4] Friebel Daniel, Mbuga Felix, Rajasekaran Srivats, et al. Structure, Redox Chemistry, and Interfacial Alloy Formation in Monolayer and Multilayer Cu/Au(111) Model Catalysts for CO2 Electroreduction [J]. The Journal of Physical Chemistry C, 2014, 118(15): 7954-7961.
[5] Hung Sung-Fu, Chan Yu-Te, Chang Chun-Chih, et al. Identification of Stabilizing High-Valent Active Sites by Operando High-Energy Resolution Fluorescence-Detected X-ray Absorption Spectroscopy for High-Efficiency Water Oxidation [J]. Journal of the American Chemical Society, 2018, 140(49): 17263-17270.
[6] Feijóo Julian, Yang Yao, Fonseca Guzman Maria V., et al. Operando High-Energy-Resolution X-ray Spectroscopy of Evolving Cu Nanoparticle Electrocatalysts for CO2 Reduction [J]. Journal of the American Chemical Society, 2023, 145(37): 20208-20213.