

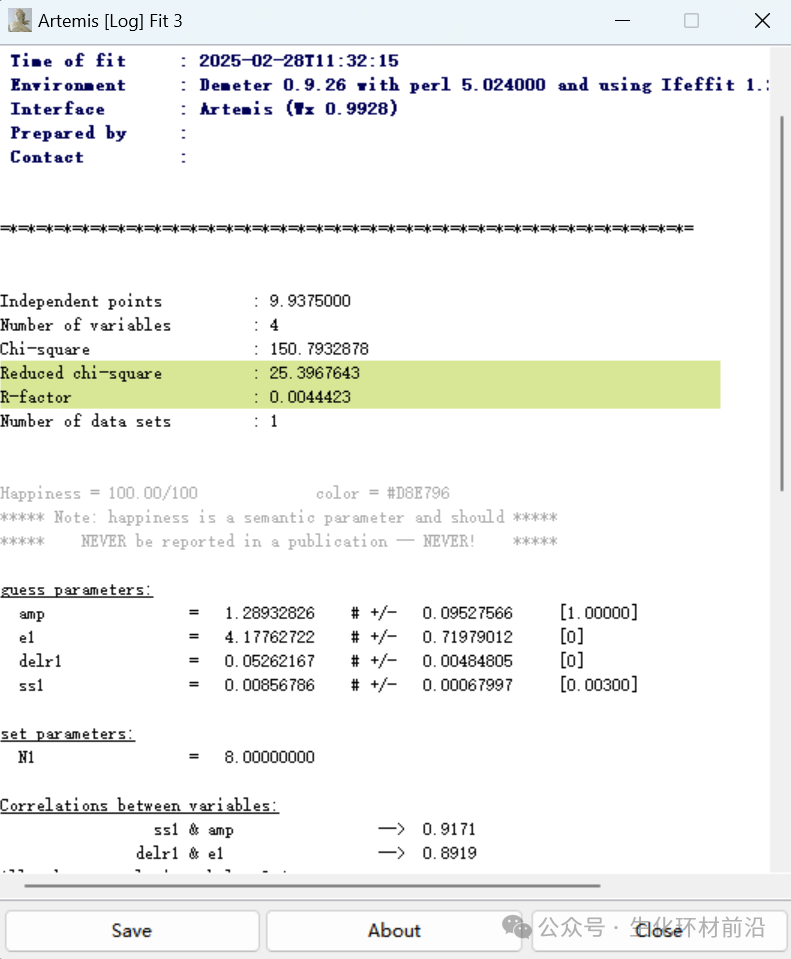
说明:本次主要推送的内容是Artemis数据拟合的基础知识,包括拟合的原理是什么?Artemis软件怎么用?利用Artemis进行EXAFS拟合的特点、流程和步骤是什么?想学更多XAFS知识请看历史内容!
Artemis对数据拟合是重点也是难点,通过拟合可以得到,配位数N、原子间距离R、无序度σ2、振幅衰减因子𝑆02、能量原点的位移E0等结构参数。
所谓曲线拟合,即将一组数据用一个带参数的的函数来模拟,调节参数直到彼此最相符合为止。在数学上意味着解一个非线性最小二乘方程的极小值问题,也就是找到最佳的一组参数或者矢量x,使得

为极小值,此处r是差值矢量,它的各分量为
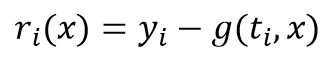
其中yi是数据点,g(ti,x)是在ti点处,拟合参数为x的拟合函数计算值。如果把f(x)作Taylor展开,以x0为中心,展至二次项
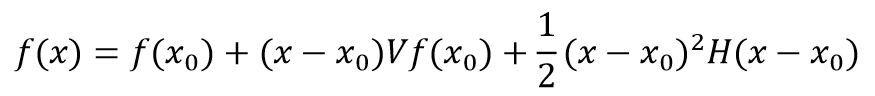
H为f(x)的二次微商,由下式给出:

其中J为Jacobi阵列

如果以函数为极小值的一点u作Taylor展开,梯度项为零,则有
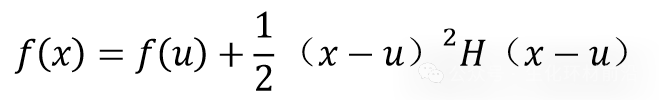
这类极小值问题,很方便地使用计算机合适的程序来解决。
对于只有一个配位壳的体系,振幅函数与相移分离的方法是适用的,不必采用曲线拟合,在Fourier变换中难以分离的两个或更多个配位壳存在时,则必须采用曲线拟合技术。
例如,两个离得很远的配位壳,铁硫蛋白中Fe-S键长的确定,或红血素中Fe-N键长的测定,都是利用曲线拟合技术来作出最后判断的。
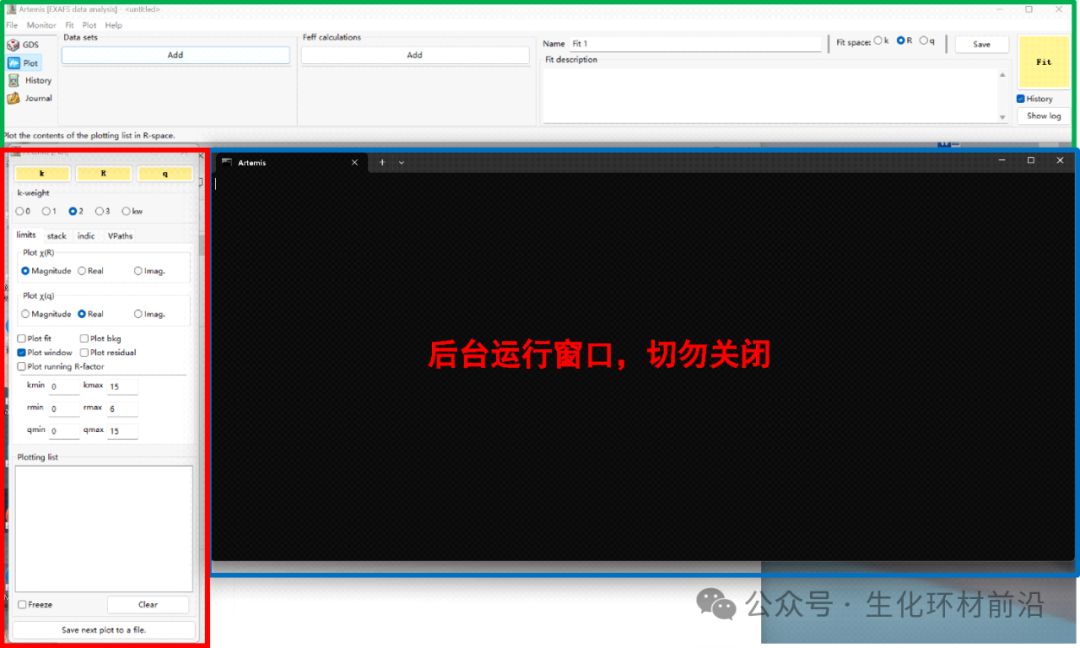
双击Artemis图标,弹出以下界面,包括黑色窗口、主窗口(上方)和绘图窗口(左侧)。注意,黑色窗口不能关闭。
在主窗口中,从左向右操作依次是:打开数据、导入模型、拟合,详细如下:
File:文件打开、保存等
Monitor:监控(所有的命令、操作)
Fit:拟合相关前置条件判定设置
Plot:绘图设置
Help:帮助文档
GDS:拟合参数设置窗口
Plot:绘图窗口
History:拟合结果窗口
Journal:日志文件窗口
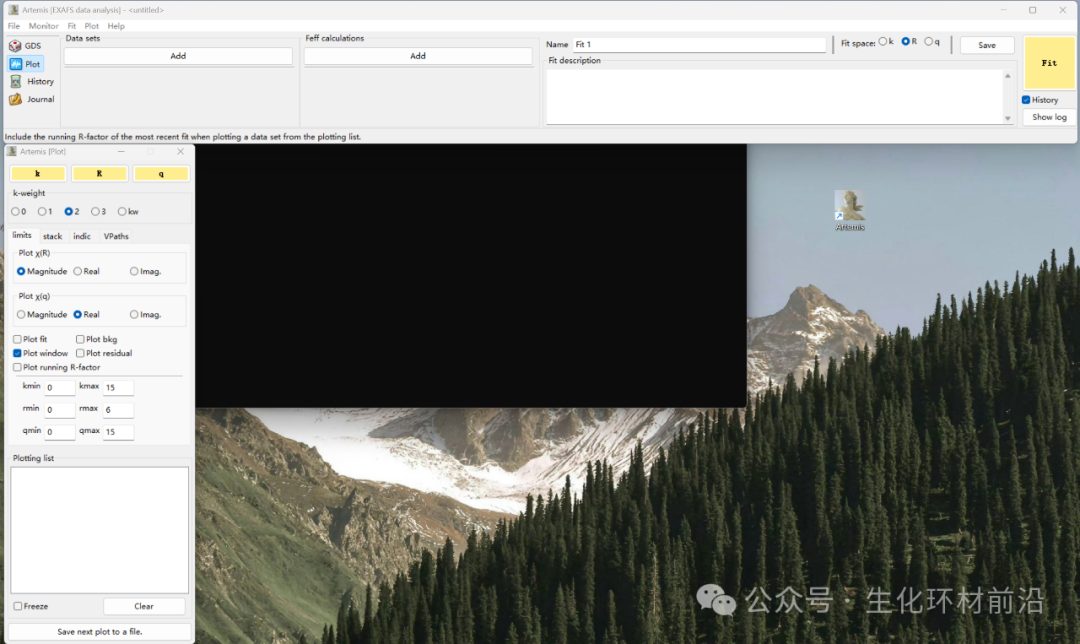

拟合参数设定窗口:设定拟合变量初始值,采用固定、限制和定义等方式,调整拟合变量的值(确认设定的参数名称正确,一一对应)。
Guess:设定初始值,不做限制。
Def:设定参数间的数学关系式。
Set:设定成固定值,不做改变。
Iguess:多数据定义。
Skip:忽略该参数,相当于程序语言中的注释符。
Restrain:设定参数限定在固定值附近。
After:设定参数间的数学关系式;
(拟合结束后,用参数的最优值代入)
绘图控制窗口:对Artemis的数据、背底、路径以及贡献、拟合结果、残差等进行绘制及绘图的控制。
绘图窗口:数据、拟合结果等的图形化显示。
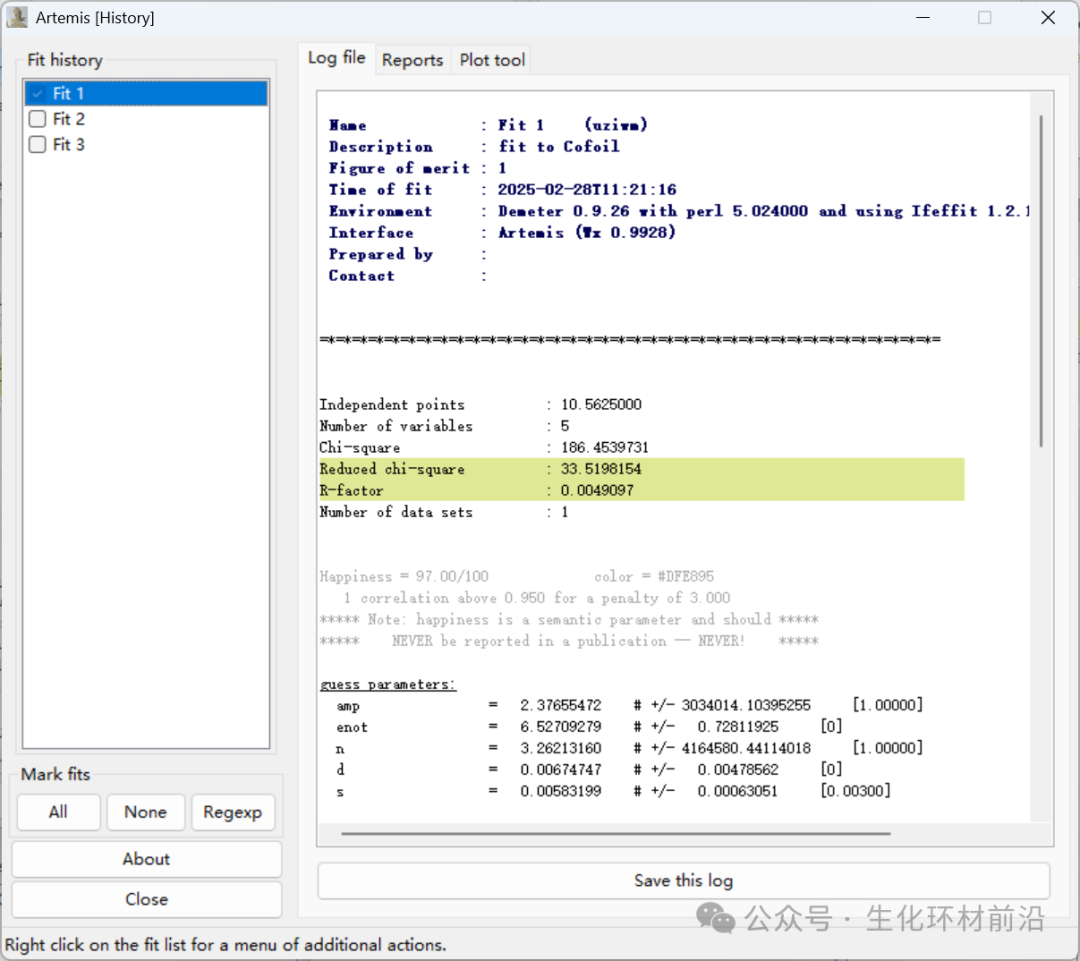
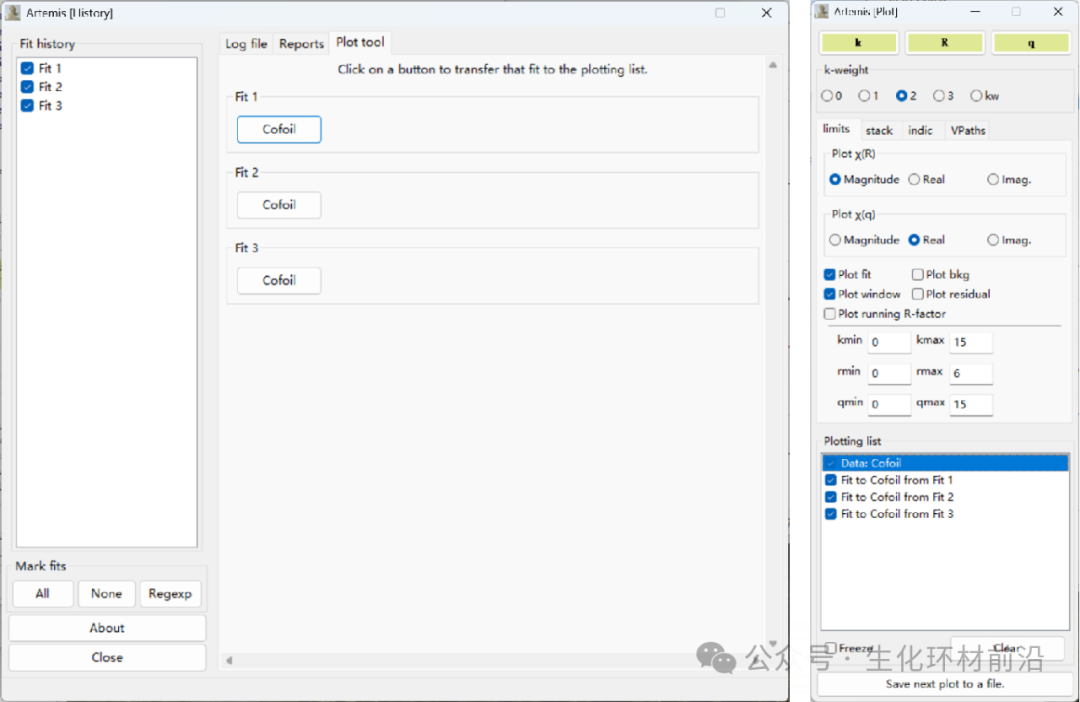
拟合历史窗口:多次拟合结果、多次拟合的统计量、拟合量的比较、添加至绘图窗口等。
Reports选项:可以选择多次拟合中的统计信息、键长、配位数等进行比较。
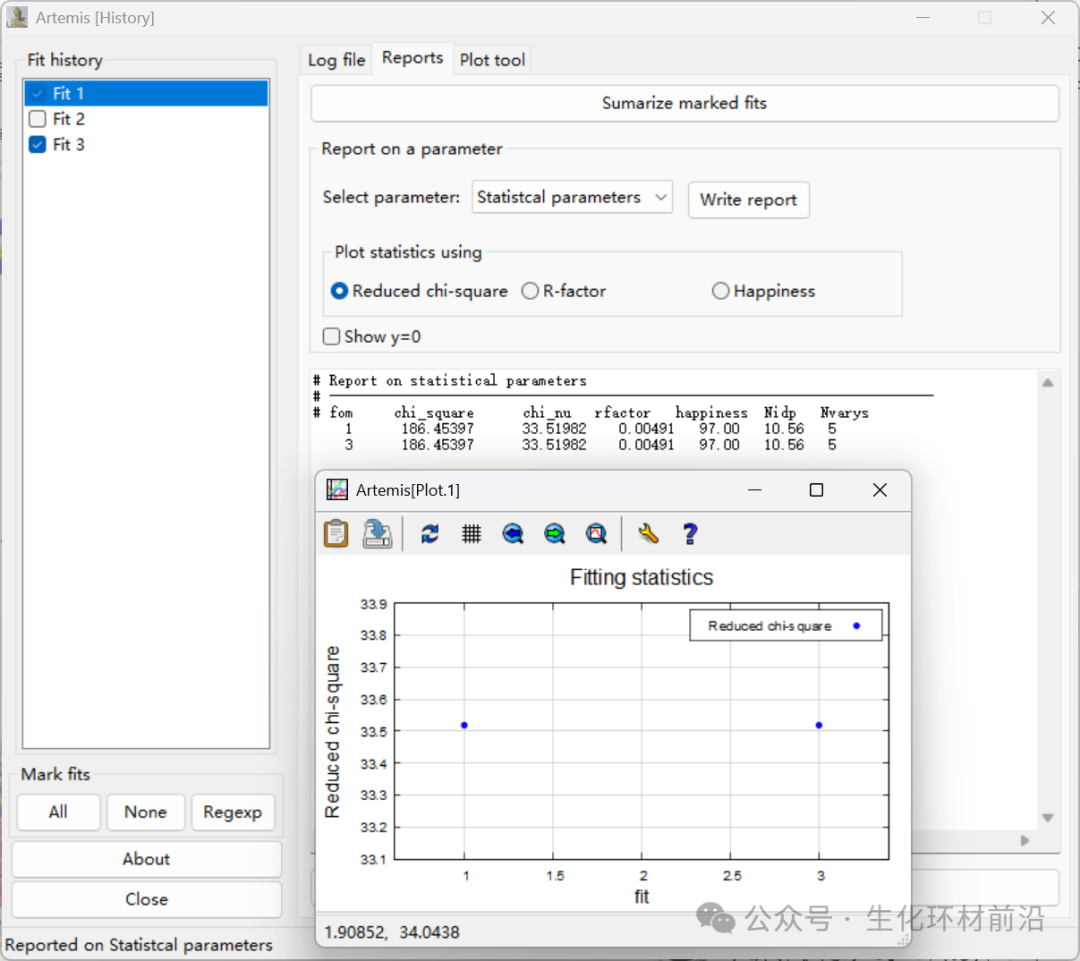
Plot tool选项:可以将多次拟合结果添加至绘图操作窗口进行比较。
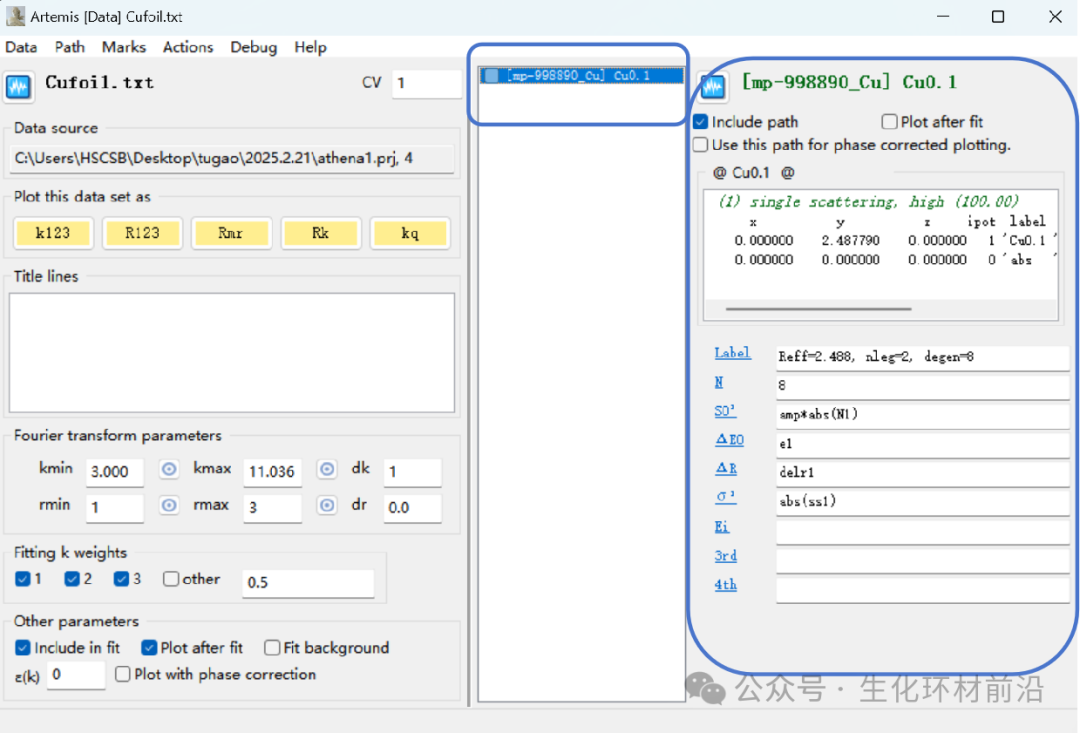
数据操作窗口:对导入数据进行各种处理,导入模型,导入路径、设定拟合参数等操作。
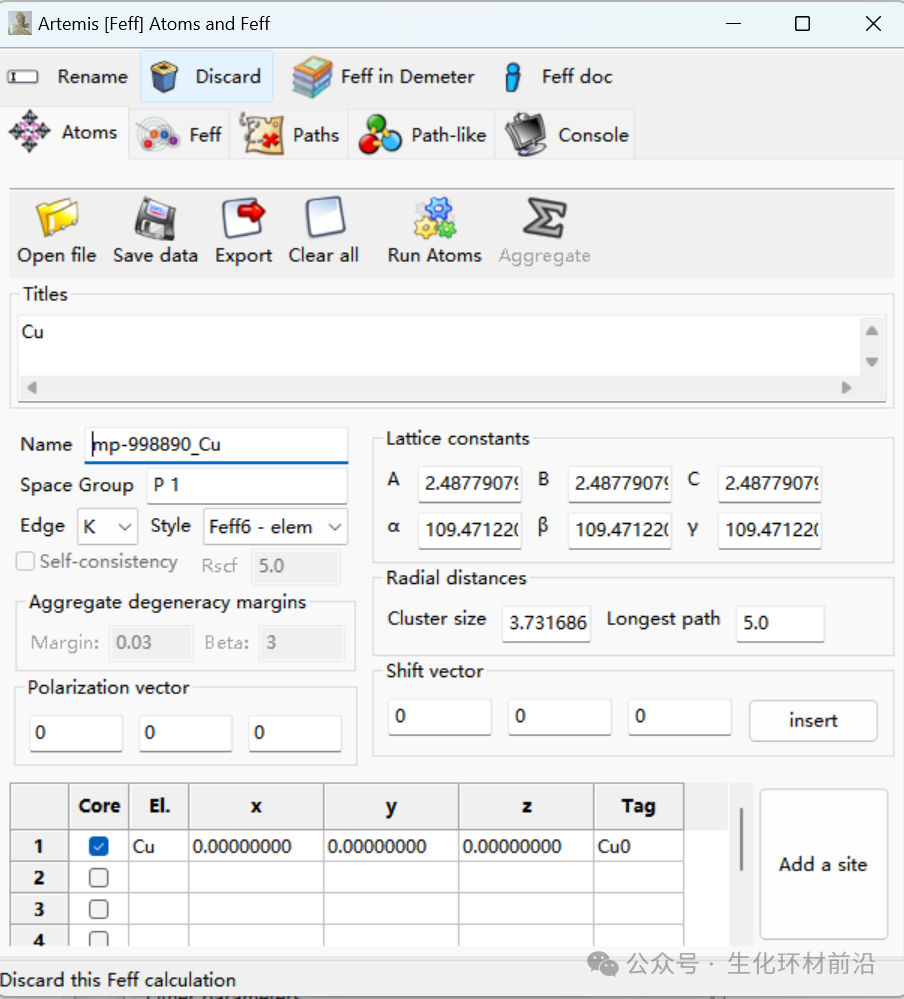
晶体学计算窗口:模型的建立、feff计算、路径导出等,相当于原ifeffit的theory选项。
拟合结果窗口:拟合得到的结果、报错等信息。
➢输入文件为不加权的EXAFS振荡函数χ(k);
➢Artemis本身不能进行EXAFS的原始数据处理(如背景扣除等);
➢基于FEFF计算的理论散射振幅和相移,对EXAFS数据进行最小二乘法非线性拟合(很容易限入局部最小,适当人为干预是必要的);
➢基本功能相当于原来UWXAFS软件包的FEFFIT程序(M. Newville),后来Bruce加入了图像用户界面(GUI);
➢内嵌有Atoms和FEFF6这2个子程序,但不包括FEFF8及更高版本。
在拟合中需要确定的参数:
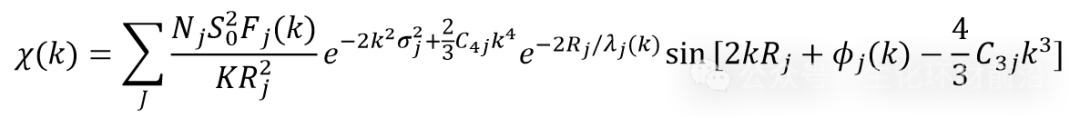
与峰强度有关:
N:路径简并度(单散射下就是配位数,coordination number)
σ2:无序度因子(Debye-Waller factor)
S02:振幅衰减因子;
C4:4阶累积量(基本不用)
与峰位置有关:
R:半程路径长度(单散射下就是原子间距离,interatomic distance)
C3:3阶累积量
∆E0:能量原点的位移
散射振幅F(k),相移ϕ(k),电子平均自由程λ(k):一般经由FEFF计算产生
拟合中,待拟合参数个数必须不大于数据能给出的独立点数。

以Cu foil数据的拟合为例
1. File→Import→χ(k)data,打开已经得到的χ(k)函数,加权,设定拟合参数。双击打开Artemis,会出现以下三个窗口。
2. 导入数据(由于上次已经使用Athena进行了数据预处理,并且保存为了.prj格式,所以这里可直接将数据拖到实验数据栏中,选中要拟合的数据再点击lmport selected data)。
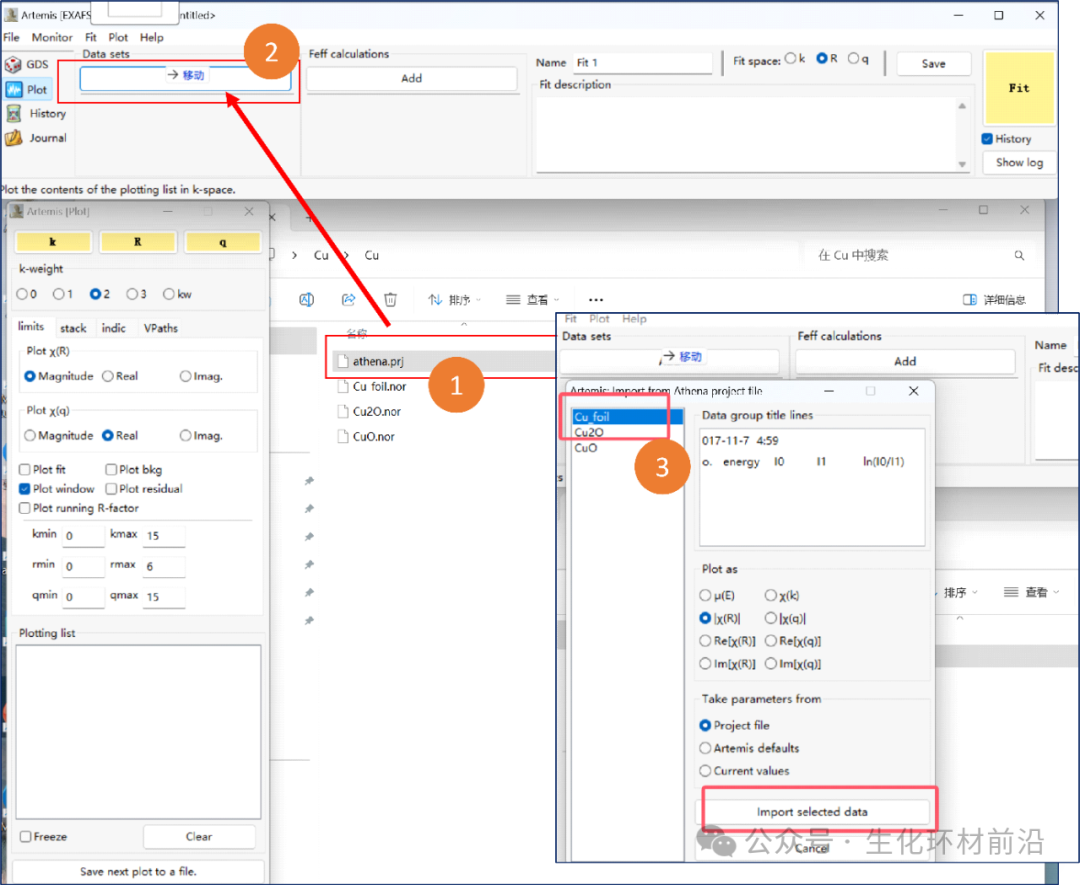
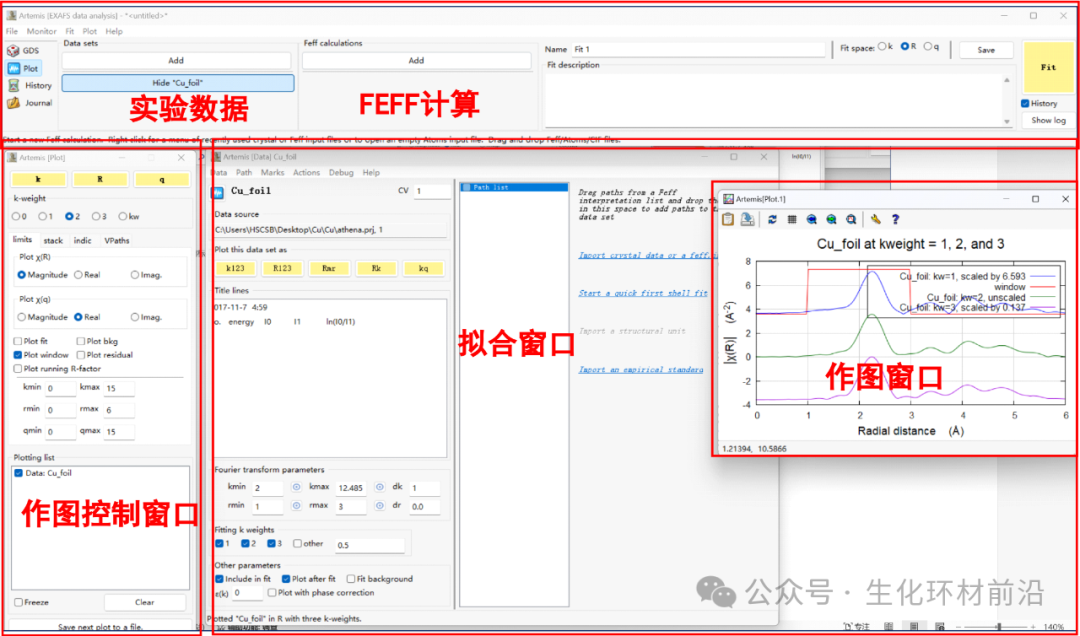
3. 数据导入后总共会出现下列窗口:实验数据、FEFF计算、做图控制窗口、做图窗口、拟合窗口、还有一个最小化的后台运行窗口。
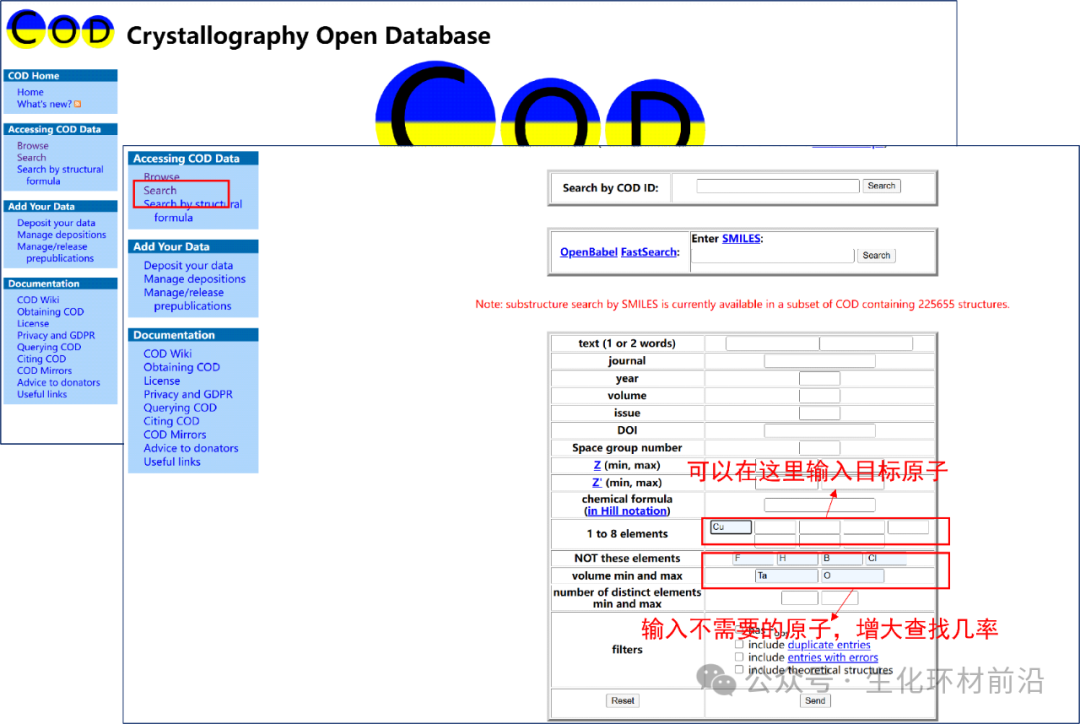
4. 接下来我们就需要导入晶体文件了,这里我是使用的Crystallography Open Database(COD)晶体库,也可以使用Materialsproject晶体库。
5. 导入晶体文件,利用Atoms模块计算原子坐标。Feff calculation→Add,打开已保存的cif文件,或者拖拽cif文件到Fef calculation 窗口再松开,或者点击“Insert crystal data or a feff.inp file”,或者根据已知的晶体结构,手工输入atoms.inp文件。
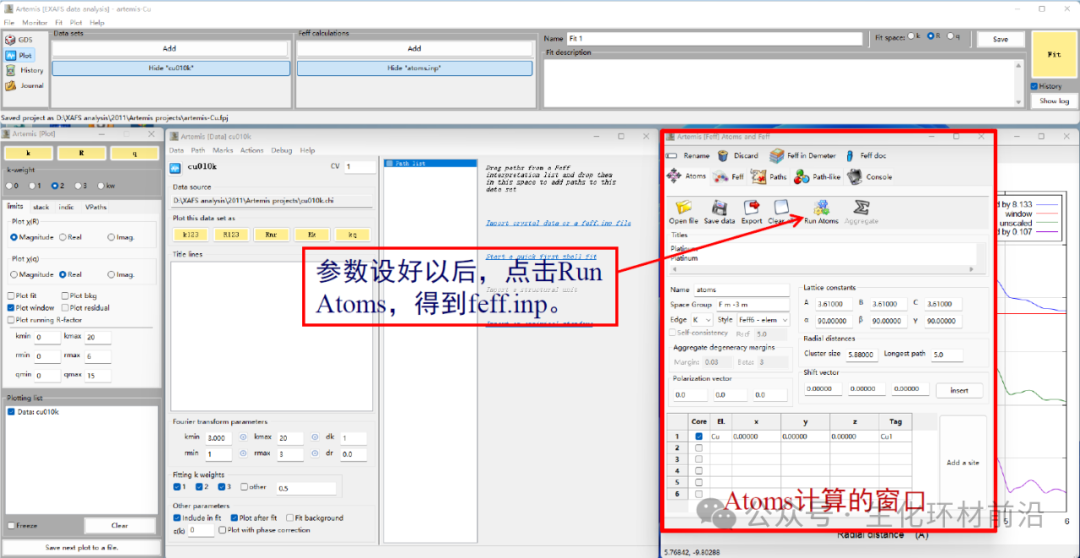
6. 利用得到的feff.inp,进行FEFF计算得到散射路径。
7. 选择Reff和待拟合的峰位置匹配(比峰位置长约0.3~0.5Å)的feff路径,将之拖拽到Path list。
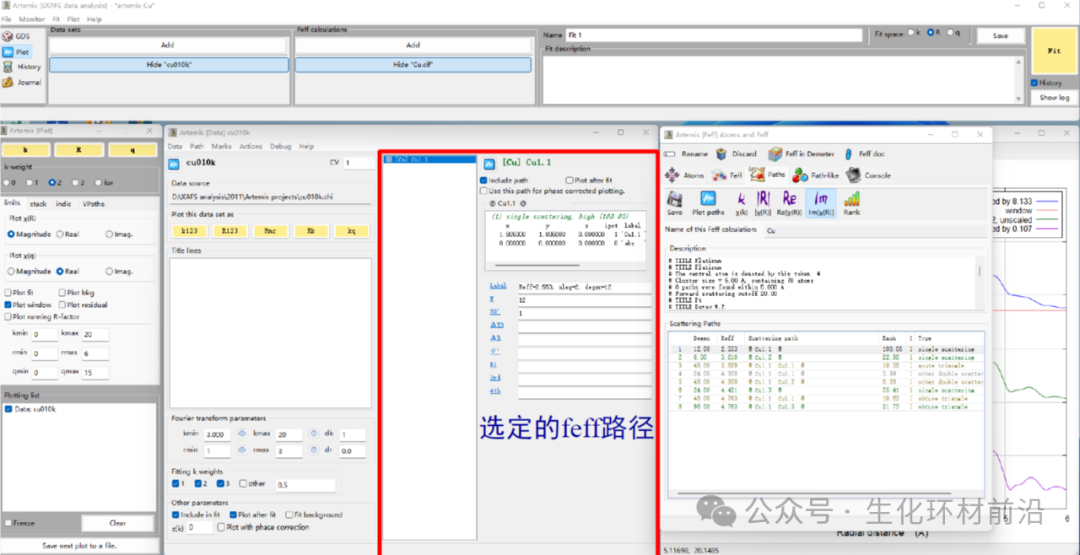
8. 设置待拟合参数。注意GDS窗口里出现的参数,必须和feff路径里用到的参数一一对应,否则会出错。
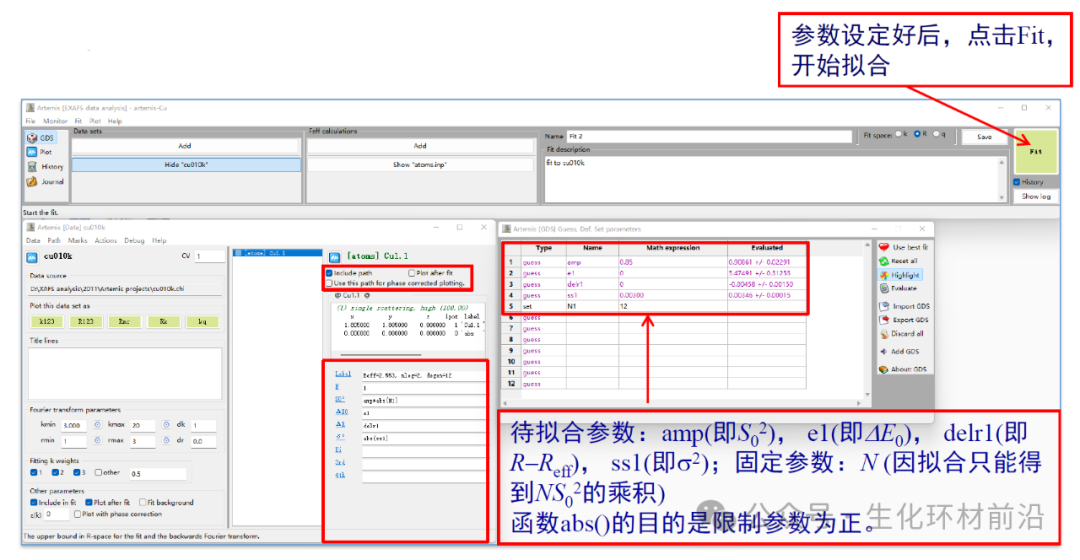
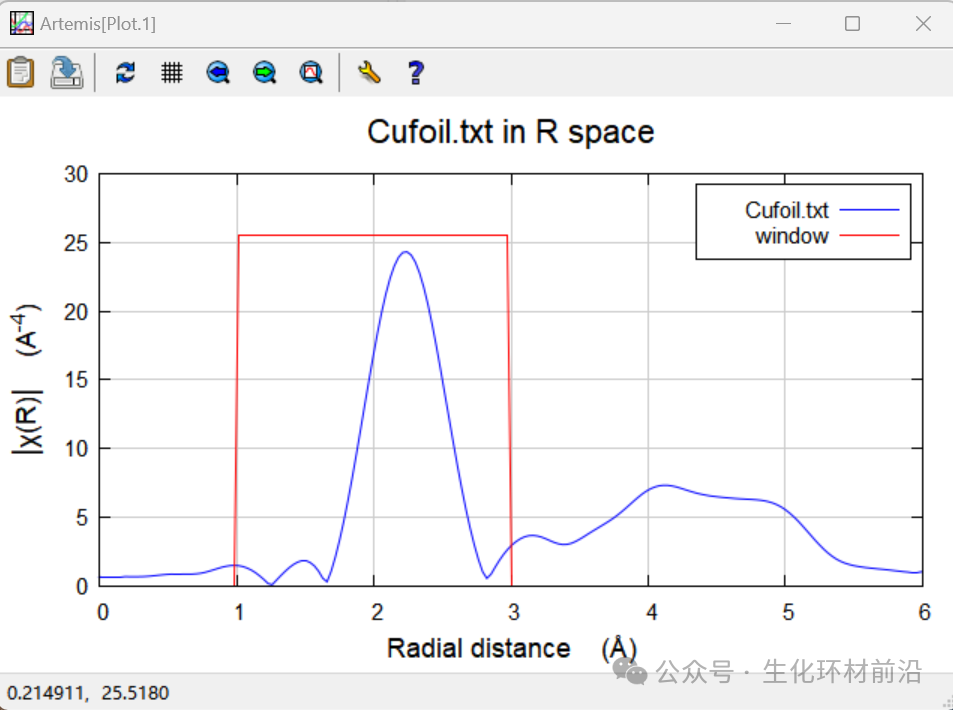
9. 查看曲线吻合情况,一般拟合范围为Δk~3-20Å-1, ΔR~1.0-3.0 Å。
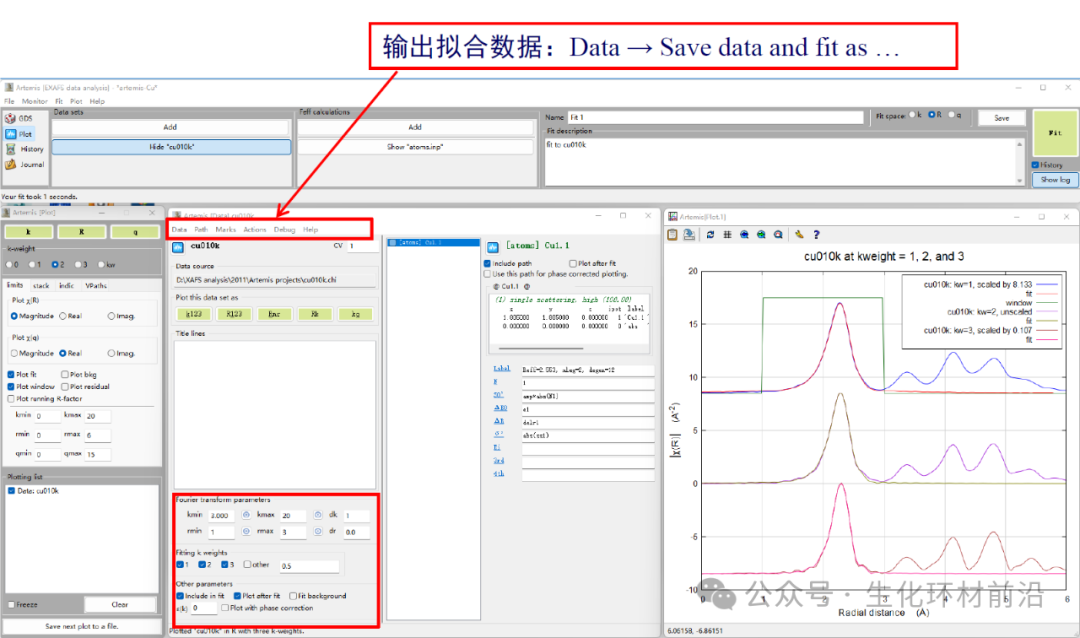
10. 查看数据报告

带有强烈主观色彩,因不同体系而异,但也仍然存在一些客观判据,如:
➢ΔE0应该在–10到+10 eV的范围,过大则不合理(其误差往往很大,误差不应作为判据);
➢同一样品中不同路径的ΔE0应相差不大,同一系列样品中同一种配位的ΔE0应接近。
➢实际拟合中,σ2合理范围为:常温下金属M-O键应该在0.0025Å2及以上;M-S键应该在0.003 Å2及以上;M-M键应该不小于其对应的foil值;同种配位更远壳层的σ2应该会大于最近邻位。
➢混合物相中,配位数是加权平均后的结果,拟合得到的配位数并不等于实际的周围原子个数;
➢待拟合参数大于独立点数Nidp=R×(2ΔR×Δk)/π
注:曲线吻合好,不代表拟合就一定可靠,必须同时得到的参数是合理的。
看完还不会,别慌,这只是基础篇,还有进阶篇,高阶篇~
Artemis对XAFS数据拟合是重点也是难点,通过XAFS拟合可以获得。