



直接电化学乙烯(C2H4)与水(H2O)的环氧化是一种可持续生产高附加值环氧乙烷(EO)的有前景的方法。然而,由于乙烯的活化缓慢以及OH中间体的形成困难,其活性仍然受限。
在此,天津大学巩金龙教授和张鹏教授等人描述了一种Ag/SnO2电催化剂的设计,以实现高效的电化学乙烯环氧化,其在膜电极组件中对环氧乙烷的法拉第效率高达39.4%,选择性达到91.5%,电流密度为25 mA/cm2。原位衰减全反射红外光谱表征结果和计算模拟表明,Ag/SnO2界面促进了乙烯的吸附和活化,生成了C2H4。同时,通过水的解离在催化剂表面生成亲电性的OH,其进一步与C2H4反应,促进了*C2H4OH的形成,从而增强了电化学环氧化活性。这项工作为通过界面工程设计用于电化学烯烃环氧化的催化剂提供了普适性指导。
相关文章以“Electrochemical epoxidation enhanced by C2H4 activation and hydroxyl generation at the Ag/SnO2 interface”为题发表在Nature Communications上!
环氧乙烷(EO)是一种重要的化学品,可用于生产乙二醇、乙二醇醚和乙醇胺,是乙烯(C2H4)的主要衍生物之一。EO的年产量超过3500万吨,近年来其需求量不断增加。目前,工业上通过在CsRe改性的Ag/Al2O3催化剂上用O2直接氧化乙烯来生产EO,该过程需要在较高温度和压力下进行(200–270°C和1–3 MPa),这可能导致显著的二氧化碳排放。为了减少碳足迹,使用可再生电力进行电加热可以替代传统的化石燃料燃烧加热系统。近年来,焦耳热、感应热、微波和等离子体等电热方法已经引起了广泛关注,并且已被用于C2H4环氧化系统。与此同时,直接C2H4电氧化是另一种具有简单性和可变性优势的重要方法,环氧化反应可能取代传统的氧气进化反应(OER),在阳极产生高附加值产品。
为了实现C2H4环氧化的高电流密度和法拉第效率,设计高效的电催化剂是关键。基于银的催化剂一直被认为是用于有氧环氧化的最活跃的工业催化剂。人们认为,银表面上适度的氧键强度对于高活性至关重要,并且亲电氧促进了环氧乙烷的形成,但C2H4在银上的吸附较弱,这会进一步阻碍电化学环氧化,正如密度泛函理论(DFT)计算所证明的那样,C2H4在Ag(111)上的结合能仅为0.06 eV。因此,设计用于以H2O进行环氧化的催化剂的关键点是促进H2O的解离以产生亲电氧物种,以及增强C2H4的吸附。
催化剂的合成与表征
通过NaBH4还原法合成了不同银(Ag)负载量的Ag/SnO2催化剂系列(即5 wt%、10 wt%和20 wt%的Ag/SnO2,分别记作5Ag/SnO2、10Ag/SnO2和20Ag/SnO2)。TEM和HRTEM用于展示不同Ag负载量样品的结构(图1a、b),在10Ag/SnO2中,SnO2载体的粒径约为100 nm,EDS映射进一步证实了不同负载量的Ag/SnO2中Ag纳米颗粒的均匀分散(图1c)。无论Ag的负载量如何,尺寸为20 nm的Ag纳米颗粒都负载在SnO2载体上。因此,与5Ag/SnO2和20Ag/SnO2样品相比,10Ag/SnO2中可以形成适量的Ag-SnO2界面。对于Ag/SnO2催化剂,Ag纳米颗粒的HRTEM图像显示出0.24 nm的条纹间距,与Ag的(111)晶面匹配。对于SnO2,晶格条纹的d间距为0.26 nm和0.33 nm,分别对应SnO2的(101)和(110)晶面(图1b)。
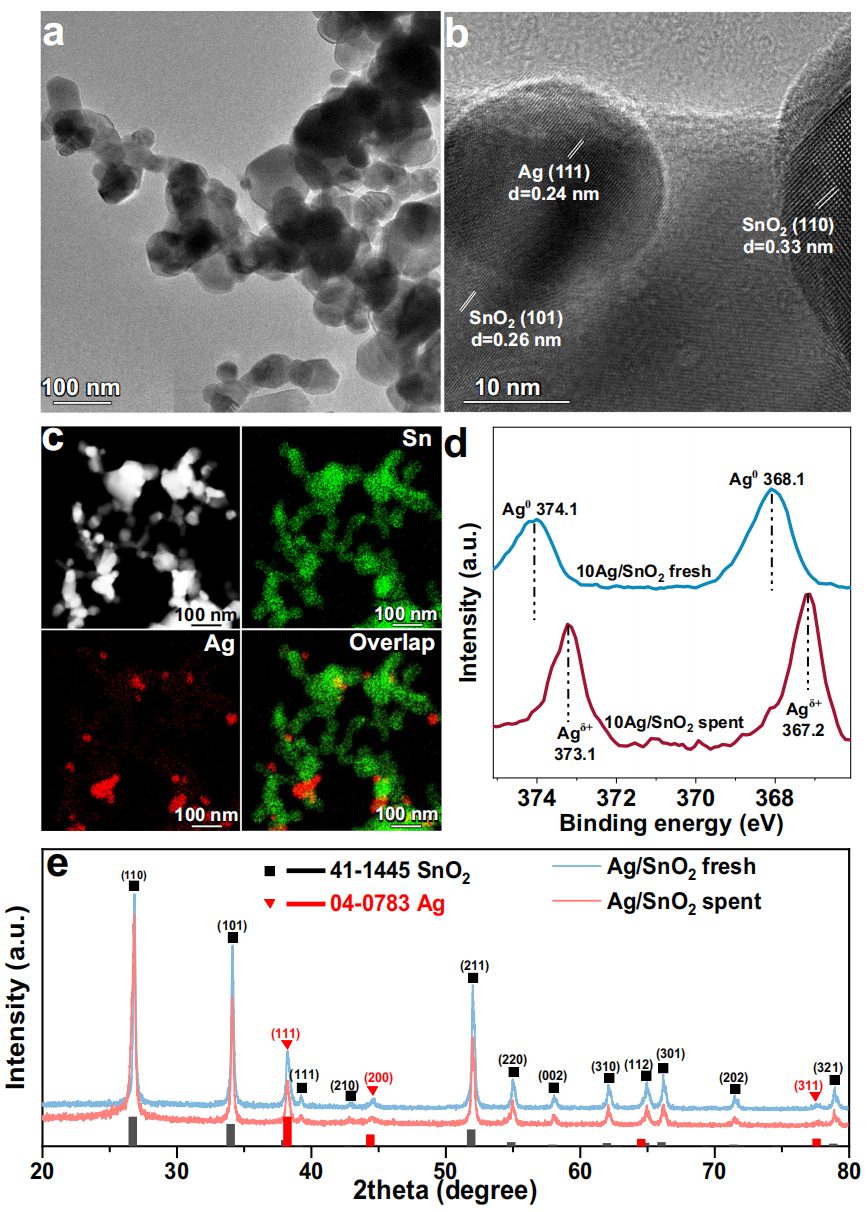
图1:10Ag/SnO2的结构表征。
Ag/SnO2催化剂的电化学环氧化性能
本研究通过线性扫描伏安法(LSV)测试了10Ag/SnO2催化剂在0.05 M KOH电解液中的电化学环氧化性能。结果显示,10Ag/SnO2在1.05 V处出现氧化峰,表明Ag氧化为AgxO。同时,10Ag/SnO2在C2H4中的电流高于在Ar中的电流,表明C2H4氧化比析氧反应(OER)更有利,且C2H4氧化的过电位为0.6 V。为了克服C2H4溶解度低的问题,采用膜电极组件(MEA)系统进行测试。1H核磁共振(NMR)和顶空气相色谱(GC)分析表明,环氧乙烷(EO)是主要产物。不同Ag负载量的Ag/SnO2催化剂中,10Ag/SnO2表现出最高的催化活性,在25 mA/cm2时实现了39.4%的EO法拉第效率(FE)和91.5%的选择性。XPS和XRD分析表明,反应过程中Ag纳米颗粒表面部分被氧化,但主体仍保持金属态。长期稳定性测试显示,10Ag/SnO2在4.2 V电池电压下保持约39%的EO FE长达8小时,随后缓慢下降至30%。反应后接触角的降低表明催化剂表面亲水性增加,可能导致C2H4反应受阻,从而引起失活。
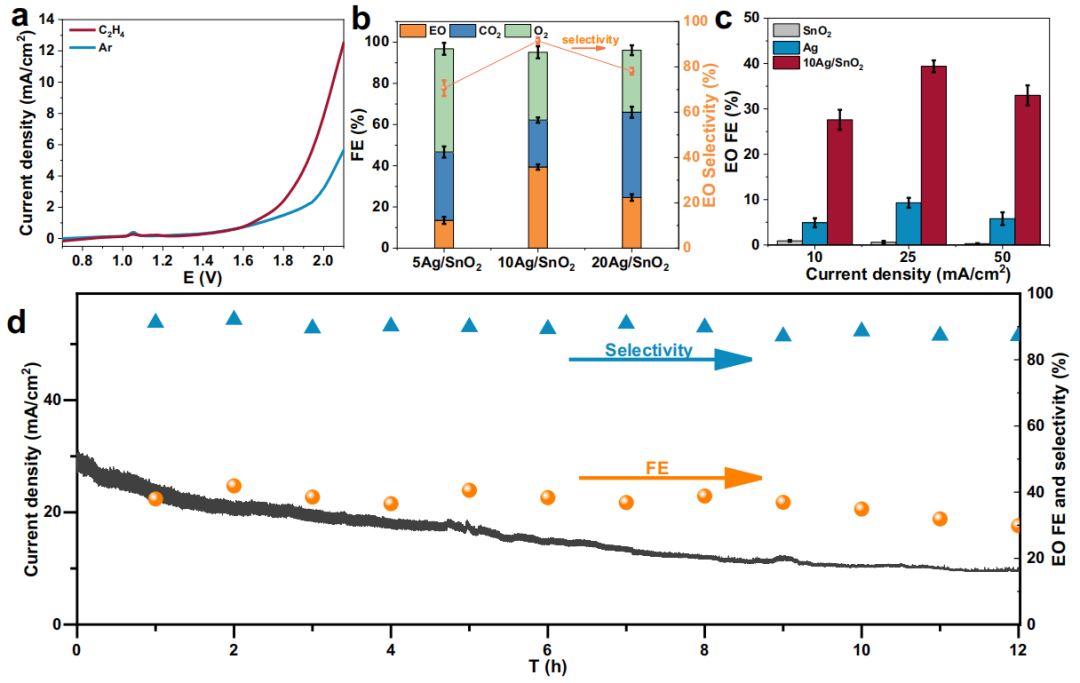
图2:C2H4的电化学环氧化性能。
C2H4在Ag/SnO2上的吸附机制研究
本研究通过红外光谱(IR)和温度程序脱附(TPD)技术,研究了C2H4在Ag/SnO2催化剂上的吸附机制。实验在室温下进行,采用预氧化的催化剂模拟电化学环氧化过程中的氧化状态。结果显示,预氧化后的10Ag/SnO2表面被氧化,但主体仍保持金属态。IR光谱表明,10Ag/SnO2对C2H4的吸附能力显著高于纯Ag和SnO2,表现为更强的C2H4特征吸收带(如3100 cm-1处的CH2不对称伸缩振动)。TPD实验进一步证实了10Ag/SnO2对C2H4的吸附能力增强,其脱附温度从Ag的-50°C提高到-6°C,并在30°C结束。这些结果表明,Ag/SnO2界面显著促进了C2H4的吸附,增加了局部浓度,有利于环氧化反应。然而,由于电化学环境的影响,C2H4的吸附行为在实际电化学环氧化过程中可能会发生变化。
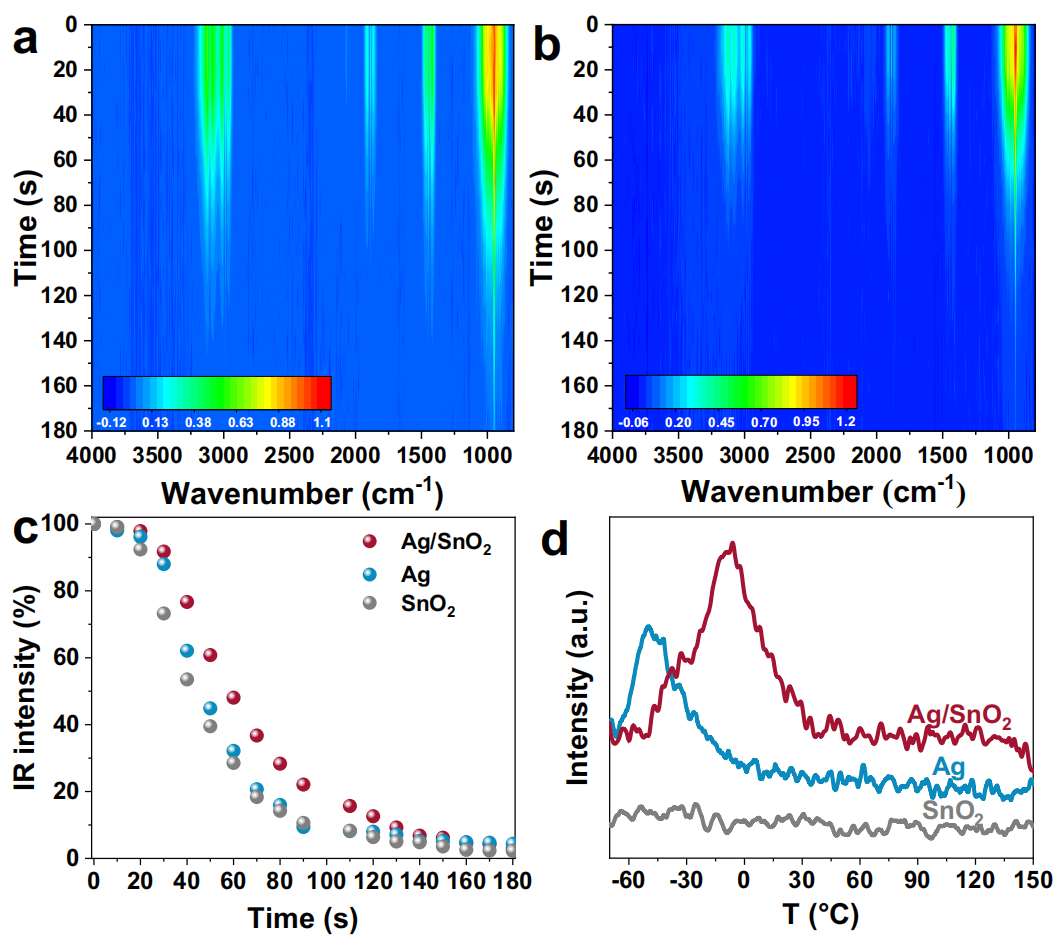
图3:C2H4吸附机理研究。
基于原位ATR-SEIRAS光谱和DFT的机制研究
本研究通过原位电化学衰减全反射表面增强红外吸收光谱(ATR-SEIRAS)和密度泛函理论(DFT)计算,深入研究了Ag/SnO2催化剂的电化学环氧化机制。ATR-SEIRAS结果显示,10Ag/SnO2在1.2 V至1.7 V的电位范围内对吸附的C2H4表现出稳定的CH2不对称伸缩振动(3100 cm-1),而纯Ag和SnO2则未观察到此吸收带,表明Ag/SnO2界面显著增强了C2H4的吸附能力。XPS和Bader电荷分析进一步揭示了Ag与SnO2之间的强相互作用,导致Ag的电子密度降低,从而促进了C2H4的吸附和活化。此外,吸附的OH在10Ag/SnO2和SnO2上于1.3 V处出现弯曲振动带(1155 cm-1),表明SnO2促进了H₂O的活化,生成了亲电性的OH,这对于环氧化反应至关重要。DEMS测试结果表明,EO的形成始于1.4 V,低于O2生成的1.5 V,表明OER中间体可能参与了环氧化反应。DFT计算进一步揭示了Ag/SnO2上C2H4环氧化的反应路径,表明通过OH中间体进行的反应在能量上更为有利。因此,C2H4在Ag/SnO2上的吸附更强,并通过SnO2产生的*OH进行氧化,实现了EO的高效生成。
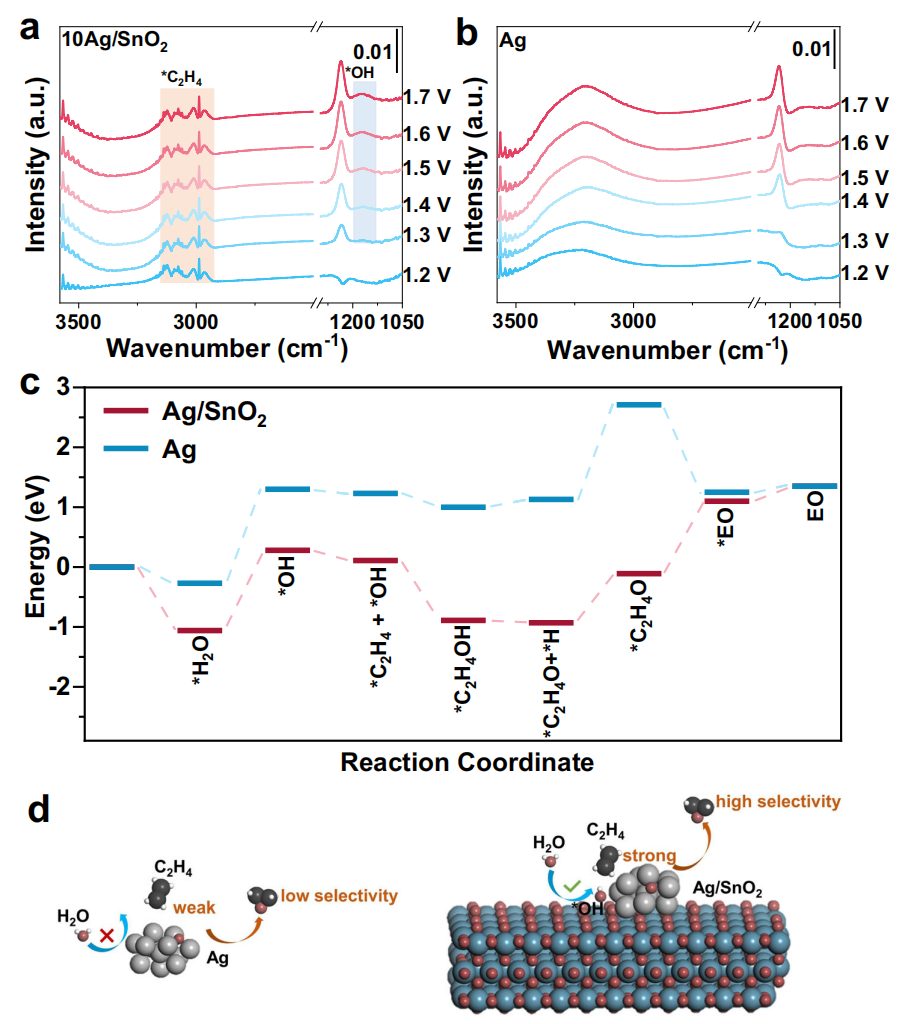
图4:Ag/SnO2的界面效应机制。
综上所述,本文设计了Ag/SnO2催化剂,用于促进C2H4的电化学环氧化生成环氧乙烷(EO)。在Ag/SnO2上观察到显著的活性提升,实现了39.4%的EO法拉第效率和91.5%的选择性。尽管EO的能量效率只能达到9.4%,但通过最小化欧姆电阻以获得卓越的伏安性能,有望提高其应用价值的能量效率。红外光谱表明,Ag/SnO2中的金属/金属氧化物界面增强了C2H4的吸附,形成了*C2H4。同时,SnO2促进了水的解离,提供了亲电性的*OH与*C2H4反应,增强了界面处的环氧化反应。本工作通过引入金属氧化物载体和支持界面的调控,为设计电化学环氧化的高性能催化剂提供了一种策略。
Hao Dong, Ran Luo, Gong Zhang, Lulu Li, Chaoxi Wang, Guodong Sun, Hongyi Wang, Jiachang Liu, Tuo Wang, Zhi-Jian Zhao, Peng Zhang*, Jinlong Gong*, Electrochemical epoxidation enhanced by C2H4 activation and hydroxyl generation at the Ag/SnO2 interface, Nature Communications, https://doi.org/10.1038/s41467-025-57223-9